Nature | 抑制脂肪酸氧化促进心脏再生
时间 : 2023-10-14心肌细胞在出生后的成熟过程中,其能量代谢从糖酵解转变为脂肪酸氧化,伴随着染色质的重新配置和退出细胞周期,这为成人心脏再生设置了障碍。随着心肌细胞的成熟,其代谢活跃性导致的氧化性DNA损伤形成了阻止心肌细胞细胞分裂的自然屏障。
-
代谢转变的影响:心肌细胞在出生后的代谢从糖酵解转向脂肪酸氧化,这一转变与心肌细胞的成熟和细胞周期退出紧密相关,为成人心脏再生设置了障碍。
-
脂肪酸氧化的中止:通过失活Cpt1b来中止心肌细胞中的脂肪酸氧化,这种代谢重编程策略可以提高心肌细胞对缺氧的抵抗力。
-
心脏再生的可能:失活Cpt1b不仅增强了心肌细胞对缺氧的抵抗,还刺激了心肌细胞的增殖,使心脏在缺血-再灌注损伤后得以再生。
-
α-酮戊二酸的关键作用:在Cpt1b突变心肌细胞中,α-酮戊二酸的积累导致了α-酮戊二酸依赖的赖氨酸去甲基酶KDM5的激活,进而影响了心肌细胞的表观遗传状态和增殖能力。
-
表观遗传与代谢的联系:这项研究揭示了心肌细胞的代谢成熟如何塑造其表观遗传景观,从而为进一步的细胞分裂创造了障碍。这为心脏再生提供了新的策略。
结论与意义
这项研究表明,代谢成熟塑造了心肌细胞的表观遗传景观,为进一步的细胞分裂创造了障碍。逆转这一过程可以修复受损的心脏。此外,αKG在染色质重新配置中的作用尚不清楚,但这项研究为其在促进心脏再生中的潜在作用提供了新的视角。这一发现不仅为我们提供了心脏再生的新策略,还为心脏疾病的治疗提供了新的思路和目标。在未来,如何利用这一新发现来设计新的治疗策略,将是一个值得进一步探讨的问题。
▉结果:
1.Cpt1b缺陷诱导心脏增生
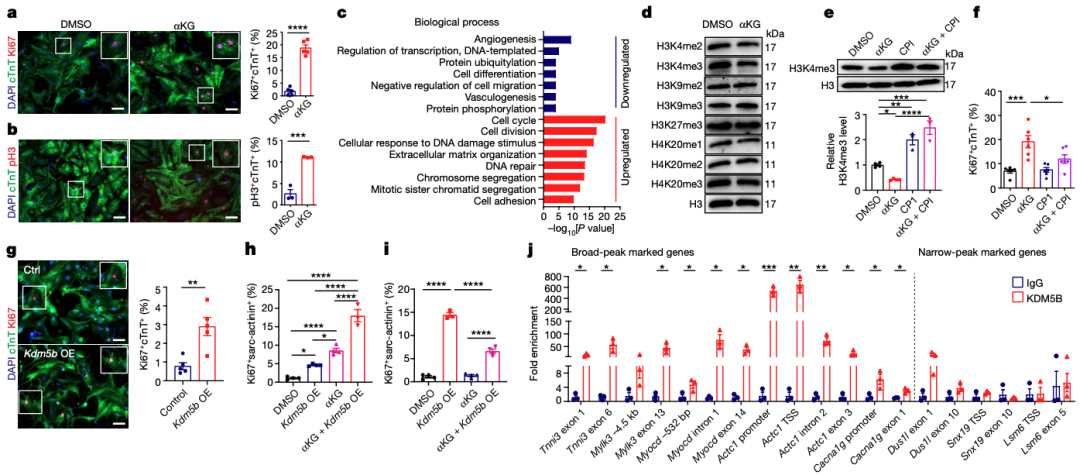
RNA测序分析发现,新生小鼠出生后第一周心肌细胞(CM)的某些基因表达发生变化,与糖酵解相关的基因下降,而与脂肪酸氧化(FAO)相关的基因上升。为了研究FAO在CM成熟中的作用,研究者使用etomoxir处理小鼠CM,结果显示增加了CM的增殖。另外,通过特定的基因操作,研究者发现失活Cpt1b基因可以刺激CM的增殖。详细的形态测量还显示,Cpt1b失活后,小CM数量增加,而大CM数量也有所上升,表明心脏的增大主要是由于细胞数量的增加,而不仅仅是细胞大小的增加。
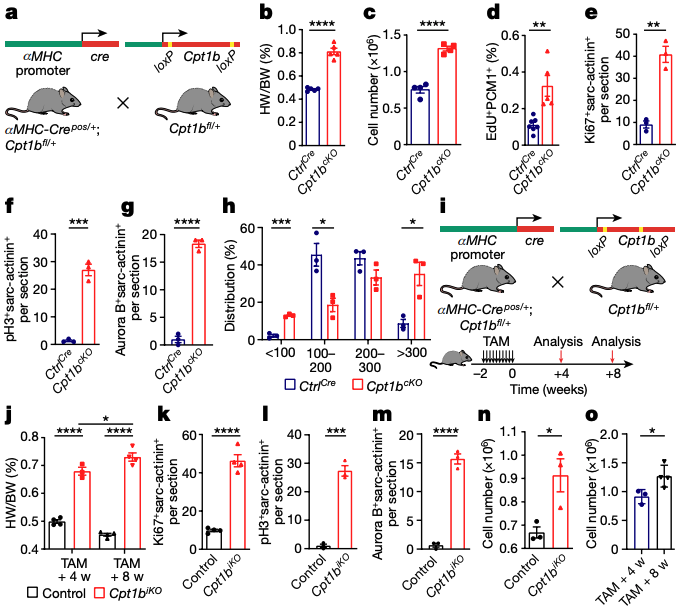
2.Cpt1b缺陷诱导心脏增生
tamoxifen(TAM)诱导的小鼠模型Cpt1biKO,Cpt1biKO小鼠的心重和心/体重比在TAM注射后4和8周显著增加。此外,Cpt1biKO小鼠的心脏显示出明显的心脏肥大和CM表面积的增加,但没有观察到纤维化或心脏功能受损。RNA测序分析显示,有1,513和1,432个基因的表达发生了显著变化。这些变化的基因主要与脂肪酸和脂质代谢过程、细胞周期进程、成熟和收缩有关。此外,电子显微镜分析和sarcomere密度的免疫荧光染色定量分析进一步证实了Cpt1b缺失CM的不成熟状态。研究者观察到抗凋亡的HIF1A信号通路的激活,这由Hif1α和其靶基因Bcl2的表达水平增加所示。与此同时,HIF1A的抑制因子Egln3的表达下调。研究者还发现,与对照心脏相比,Cpt1b缺失心脏中的γH2A.X+心脏细胞核的百分比显著下降,这表明抑制FAO可以减少DNA损伤或刺激DNA修复。
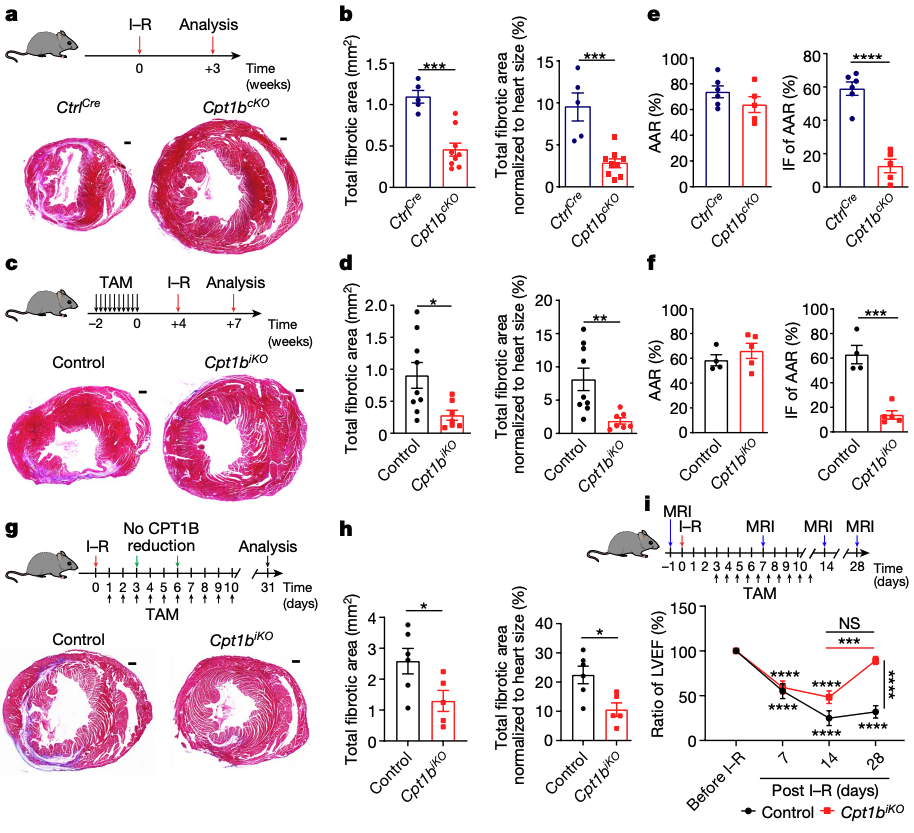
3.阻断FAO促进心脏再生
对Cpt1bcKO和Cpt1biKO小鼠进行了缺血-再灌注(I-R)损伤实验,3周后Cpt1bcKO和Cpt1biKO小鼠的I-R诱导的瘢痕几乎消失。这种几乎完全的瘢痕形成预防伴随着大量小的、圆形的Ki67+和aurora B+ CMs的出现,特别是在Cpt1b缺失心脏的边界区域。研究者在I-R损伤后24小时内在两种突变心脏中检测到了减少的梗死区域,并在I-R损伤后48小时内在Cpt1bcKO小鼠中检测到了更好的心脏收缩,这表明抑制FAO也可以对I-R损伤提供保护。
与这些发现一致,与对照CM相比,Cpt1b缺失CM的细胞死亡率显著降低。为了确定CM增殖对I-R损伤后心脏再生的贡献,并排除增强保护作为瘢痕形成缺失的主要原因,研究者在应用I-R损伤后1天开始删除Cpt1b。Cpt1biKO动物的纤维化瘢痕区域显著减少。观察到I-R手术后4周的心脏功能大大恢复,几乎达到了受伤前的同一水平,这明确地表明Cpt1b失活促进了心脏再生。得出结论增强的CM增殖和心脏保护都有助于在FAO缺乏的小鼠中冠状动脉闭塞后减少瘢痕形成。
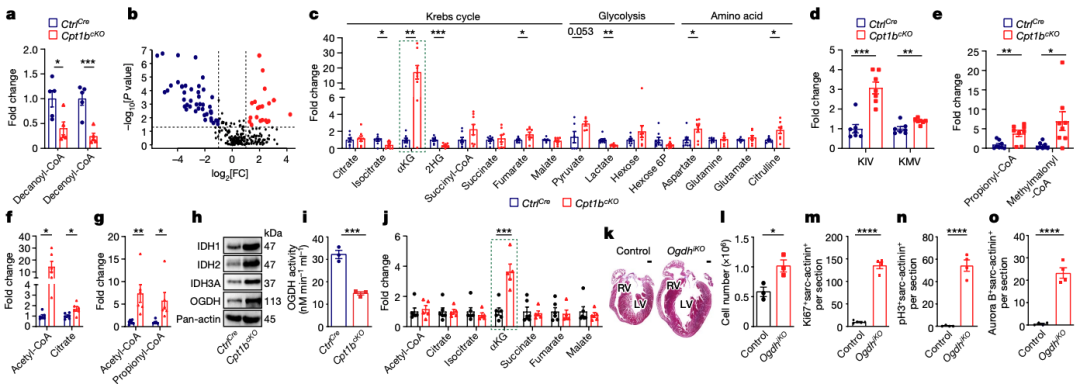
3.Cpt1b 缺失会增加 CM 中的αKG 水平
研究者探讨了阻断FAO如何重新规划心肌细胞(CMs)的新陈代谢。结果显示,Cpt1bcKO CMs中长链脂肪酸的利用受到了抑制,而中链或短链脂肪酸的代偿性利用并不明显。尽管FAO受到抑制,但CMs中的乙酰-CoA和大多数克雷布斯循环代谢产物的水平并没有显著变化,这意味着CMs采用了不同的能量生产方式。研究者推测,增加的葡萄糖氧化可能在一定程度上补偿了Cpt1b缺失CMs中的FAO损失。此外,氨基酸可能是Cpt1b-depleted CMs的另一个能量来源,因为研究者检测到了与氨基酸代谢有关的代谢物的主要富集。值得注意的是,研究者在Cpt1b失活后的CMs中检测到了αKG的显著增加,几乎增加了20倍。此外,与对照CMs相比,Cpt1b-deficient CMs中的OGDH酶活性减少,尽管OGDH复合物组分的蛋白质浓度增加。总的来说,增加的合成和减少的代谢作用协同导致了FAO中止后CMs中αKG的积累。
4.CM 特性基因中的 H3K4me3 减少
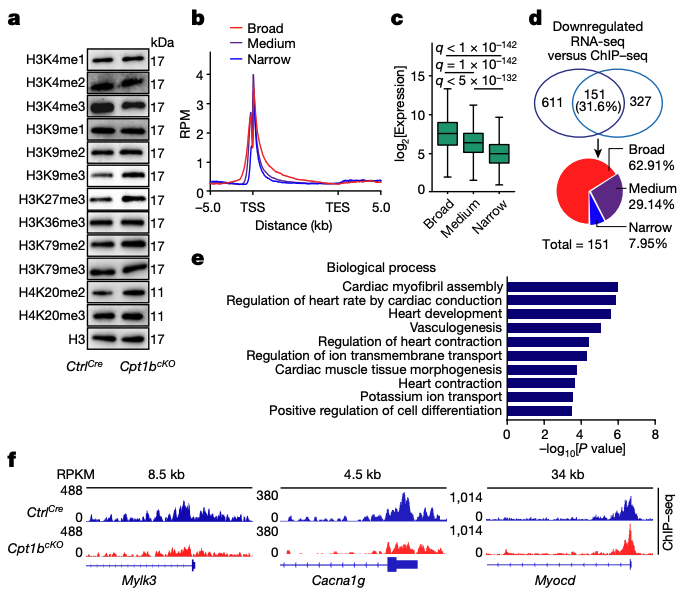
5.活化的 KDM5 可刺激增殖