首页
业务介绍
▼
SCI评估
SCI编译
实验外包
数据统计
课题申请辅导
信誉保障
SCI期刊
联系我们
Science:人NLRP1炎症小体活化新机制
时间 : 2022-07-16
机体固有免疫系统可以通过保守的感应蛋白质来识别病原体相关的分子模式
(pathogen-associated molecular patterns)
和危险信号相关的分子模式
(damage-associated molecular patterns)
。含NACHT, LRR和PYD结构域蛋白
(NACHT, LRR, and PYD domain-containing proteins,简称NLRPs)
可以装配形成炎症小体这一结构,用以响应胞内的病原体和危险信号,从而导致以caspase-1活化,gasderminD孔结构形成,以及IL-1分泌释放等为特征的细胞胶亡
。人类NLRP1炎症小体之所以备受关注,主要是基于其结构域排列特殊,并且与啮齿类同源蛋白具有一定差异
。蛋白酶DPP8和DPP9的抑制剂,比如Val-boro-Pro
(简称VbP)
,是目前唯一已知可以同时活化人类和啮齿类NLRP1炎症小体的分子
,还包括一个人类炎症小体相关感应蛋白CARD8
。近期工作显示人类NLRP1可以感应双链病毒RNA、病毒蛋白酶
,UVB辐射
【15】
等等。而这些均不能活化啮齿动物NLRP1炎症小体。
人类NLRP1主要表达于皮肤和呼吸道上
【12】
。其突变或者是单核苷酸多态性均有可能导致皮肤功能紊乱。因此,人类NLRP1在皮肤免疫方面具有独特的作用。UVB波长在280到315纳米之间,是造成灼伤的主要因素,也是目前为止NLRP1激动剂中与皮肤最为相关的
【15】
。但是,NLRP1感应UVB的分子机制仍不清楚。2022年7月14日,来自新加坡南洋理工大学的
Franklin L. Zhong
研究组在
Science
上发表题为
ZAKa-driven ribotoxic stress response activates the human NLRP1 inflammasome
的文章,
确定ZAKa/MAPK20驱动的应激响应可以活化人NLRP1炎症小体。
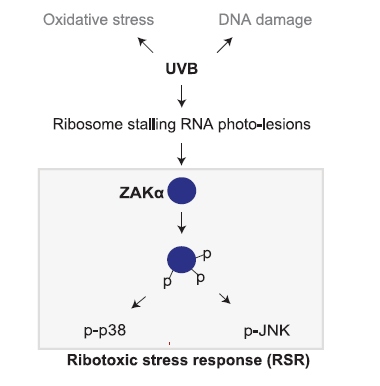
首先,作者通过永生化角蛋白形成细胞系N/TERT-1确定,单一的DNA损伤和自由基氧化损伤均不能驱动UVB诱导的NLRP1炎症小体活化。因此,作者猜测活化NLRP1炎症小体的可能是UVB驱动的RNA损伤。作者通过用核苷酸类似物4-SU预处理细胞,用以提高RNA对UVA的敏感性。并发现RNA的光损伤很可能是UV诱导NLRP1炎症小体活化的的上游信号。
近期有报道指出,UVB驱动的SAPK活化依赖于ZAKa激酶。活化的ZAKa可以进一步自磷酸化和遴选话其下游的SAPK。这一通路也被称之为核糖毒性应激响应
(ribotoxic stress response,简称RSR)
。作者确定,RSR和NLRP1炎症小体活化依赖于ZAKa的核糖体结合与RSR感应结构域。
接下来,作者研究ZAKa激活毒素茴香霉素
(anisomycin,简称ANS)
和四环霉素
(doxyvinenol,简称DON)
的具体作用。作者发现,这些ZAKa激动剂同样也可以活化NLRP1炎症小体,且其活化机制并不是通过影响翻译作用。
再下来,作者在NLRP1敲除细胞系中构建和转入了人NLRP1, NLRP1敲PYD, 鼠NLRP1B
(muNLRP1B)
,或人 CARD8等片段。作者发现,ANS等仅能活化过表达NLRP1, NLRP1敲PYD的NLRP1炎症小体。人NLRP1具有独特的N端延长结构域NLRP1
DR
,而鼠NLRP1和CARD8并没有。作者通过敲除NLRP1
DR
确定,这一结构域是ZAKa驱动的NLRP1炎症小体活化的必须条件。
另外,作者还发现细胞经UVB或ANS处理后,NLRP1
DR
-GFP条带会出现明显迁移。作者确定这是NLRP1的NLRP1
DR
片段发生明显的高度磷酸化。作者通过对这一片段上的丝苏氨酸位点进行研究发现,单一磷酸化位点突变足以调控ZAKa驱动的NLRP1炎症小体活化。
ZAKa可以活化多种SAPKs,最后,作者研究其他激酶是否也参与ZAKa驱动的NLRP1炎症小体活化。作者通过多种激酶的抑制剂发现,p38同样参与这一过程。ZAKa通过磷酸化NLRP1起始其活化,而p38可以扩大和加强这一响应。
综上所述,作者
发现人NLRP1可以感应UVB以及毒素诱导的核糖毒性应激响应。
此响应可以导致NLRP1人特异性结构域NLRP1
DR
依赖ZAKa激酶以及下游p38的高度磷酸化,突变NLRP1
DR
单一磷酸化位点就可以去除UVB和核糖毒性驱动的细胞胶亡。上述结果
说明了人皮肤角蛋白形成细胞可以感应UVB,展示了集中人NLRP1特异性激动剂,并且建立了以RSR为核心的炎症小体驱动细胞凋亡机制。