脓毒症(sepsis)是一种危及生命的疾病,定义为对感染的全身免疫和炎症反应失调引起的多器官功能障碍。由于炎症的发生与血栓的形成密切相关,脓毒症中的过度炎症反应通常与增强的微血管血栓形成和失调的凝血级联反应相关。此外,血小板在脓毒症中发挥重要作用,因为脓毒症患者中经常观察到血小板计数降低,表明在脓毒症发展过程中微血管血栓形成部位发生血小板的隔离和消耗。而且研究表明较低的血小板计数与脓毒症患者的较高死亡率密切相关。血小板还调节对感染的免疫应答,并有助于炎症和病原体消除,使其参与脓毒症的全身炎症。
核苷酸寡聚化结构域(NOD)样受体(NLR)是一类模式识别受体(PRR),其感测病原体相关分子模式(PAMP)和损伤相关分子模式(DAMP)以刺激炎症反应作为响应感染的第一道防线。在感测 PAMP 或 DAMP 后,NLRs 可以形成超分子复合物,称为炎性小体,通过加工 pro-IL-1β 或 pro-IL-18 使 IL-1β 或 IL-18 成熟以调节免疫或炎症反应。NOD 样受体家族pyrin结构域6(NLRP6)主要在粘膜组织中表达并维持肠道微生物群稳态。鉴于肠源性细菌易位是脓毒症病理生理学的关键驱动因素,NLRP6 根据不同组织参与宿主对病原体的防御,作为促炎或抗炎作用,表明 NLRP6 的细胞类型或组织特异性功能。然而,NLRP6 在脓毒症中的细胞类型特异性作用仍然知之甚少。
近日,徐州医科大学乔建林教授团队在 Blood 上刊发了题为 Platelet NLRP6 protects from microvascular thrombosis in sepsis 的研究文章。该文章通过血小板特异性 NLRP6 敲除小鼠和败血症的盲肠结扎穿孔模型证明了血小板 NLRP6 的缺失增加了死亡率,增强了肺和肝脏中的微血管血栓形成,并促进了败血症后的血小板活化、血小板-中性粒细胞相互作用以及中性粒细胞胞外陷阱(NETs)的形成。进一步发现 NLRP6 促进了活化的血小板中 TRIM21 和 TAB1 之间的相互作用,导致 TAB1 的 K48 连接的多泛素化以及随后的降解。该研究确定了血小板 NLRP6 在败血症微血管血栓形成过程中的新型保护作用,暗示其作为治疗败血症的新型靶点。
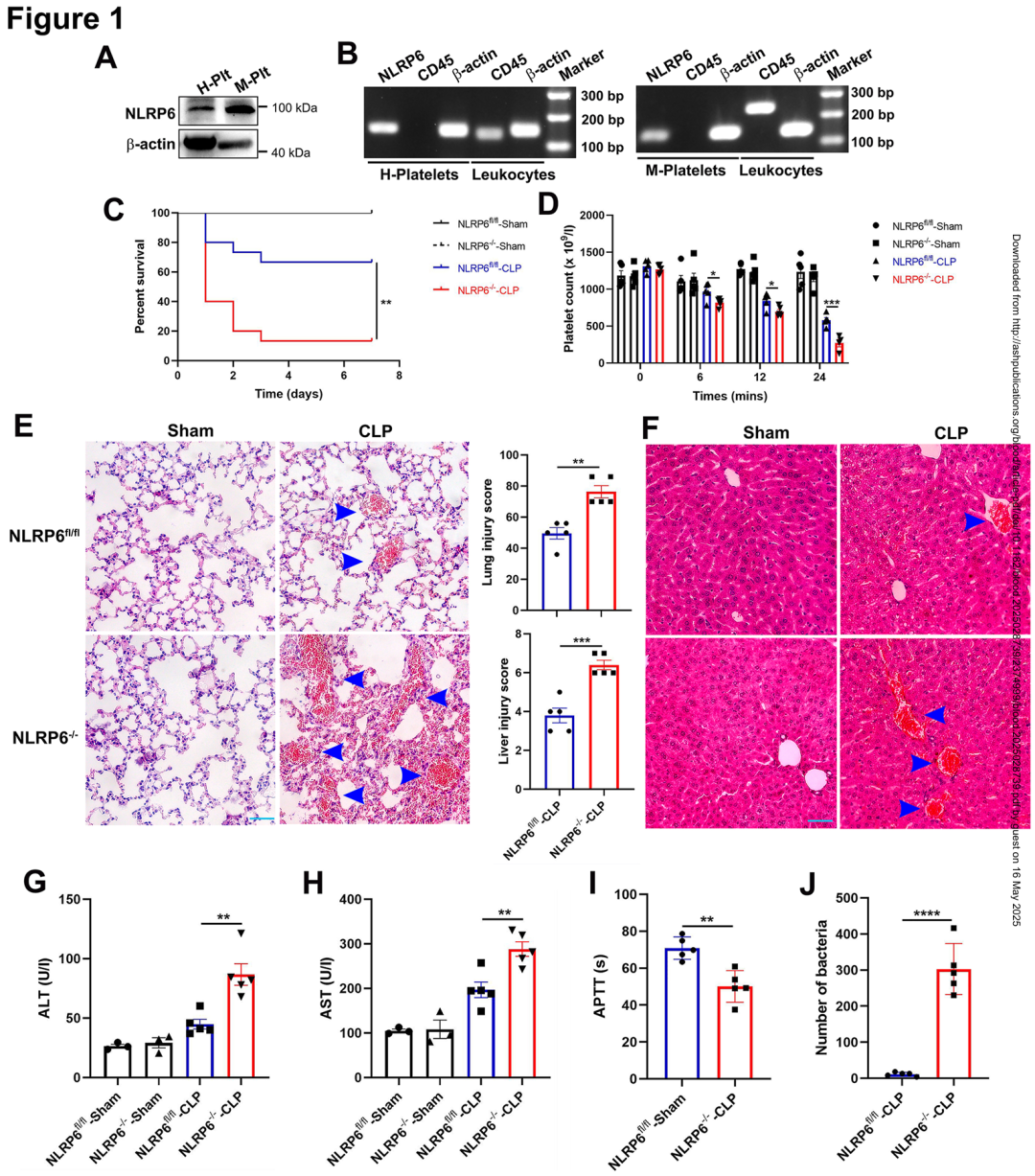
01.血小板NLRP6缺陷增加脓毒症患者微血栓形成并降低生存率
作者首先通过蛋白质印迹(Western blot)和逆转录聚合酶链反应(RT-PCR)首次证实 NLRP6 在人和小鼠血小板中特异性表达,且不受白细胞污染干扰(CD45阴性)。为探究其功能,研究团队构建了巨核细胞/血小板特异性 NLRP6 敲除小鼠(NLRP6⁻/⁻),并通过盲肠结扎穿刺(CLP)模型模拟败血症。结果显示,NLRP6⁻/⁻ 小鼠在败血症后72小时存活率仅为13%,显著低于对照组(NLRP6ᶠˡ/ᶠˡ小鼠的40%)。外周血小板计数在24小时后下降37%(从6.5×10⁵/μL降至4.1×10⁵/μL),提示血小板消耗加剧。
组织病理学分析显示,NLRP6⁻/⁻小鼠肺组织中微血栓面积增加2.3倍(H&E染色显示肺泡壁血管扩张充血,血栓密度为12.8个/mm² vs. 5.6个/mm²),肝脏出现广泛微血栓并伴随肝细胞肿胀,血清 ALT 和 AST 水平分别升高1.8倍(ALT: 128 U/L vs. 70 U/L)和1.5倍(AST: 95 U/L vs. 63 U/L)。凝血功能检测显示活化部分凝血活酶时间(APTT)缩短16%(28秒 vs. 33秒),且血液细菌载量增加3.2倍(CFU计数:1.2×10⁶ vs. 3.7×10⁵)。这些结果首次揭示血小板 NLRP6 在败血症中通过抑制微血栓形成和细菌扩散发挥保护作用。
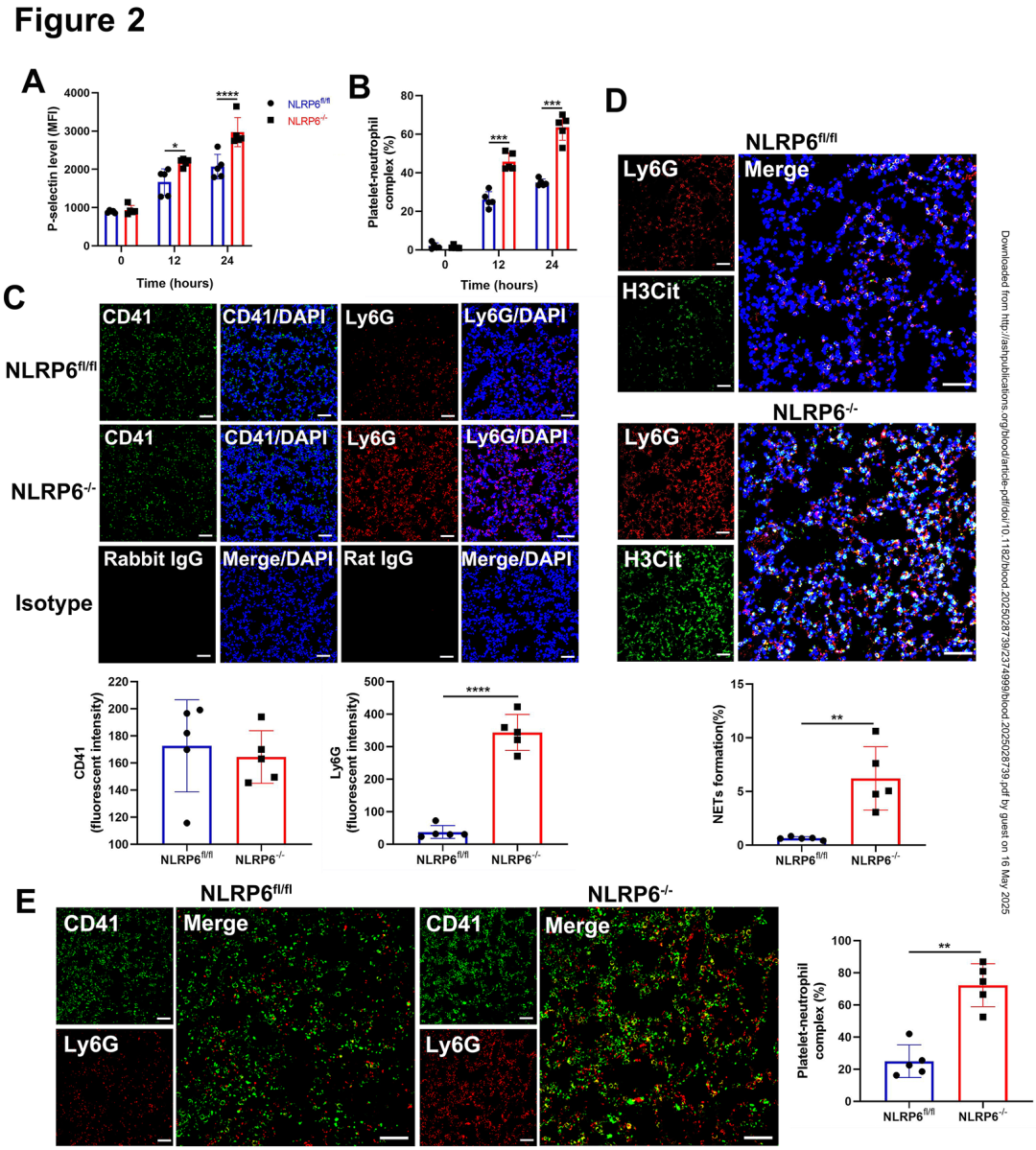
02.NLRP6缺失增强血小板-中性粒细胞互作及NETs形成
为解析 NLRP6 调控败血症微血栓的机制,作者通过流式细胞术发现 NLRP6⁻/⁻ 小鼠外周血血小板激活标志物P-选择素(CD62P)表达量在败血症后24小时升高52%,血小板-中性粒细胞复合物(PNAs)增加1.9倍。肺组织免疫荧光显示,NLRP6 缺失虽未显著改变血小板(CD41⁺细胞)浸润,但中性粒细胞(Ly6G⁺细胞)浸润增加2.1倍,且中性粒细胞胞外诱捕网(NETs,H3Cit⁺区域)面积扩大3.7倍。透射电镜进一步显示 NLRP6⁻/⁻ 血小板α颗粒和致密颗粒数量减少30-40%,但膜表面受体(如αIIbβ3、GPIb、GPVI)表达无差异。这些结果提示NLRP6通过抑制血小板过度激活及与中性粒细胞的异常互作,限制NETs驱动的微血栓形成。
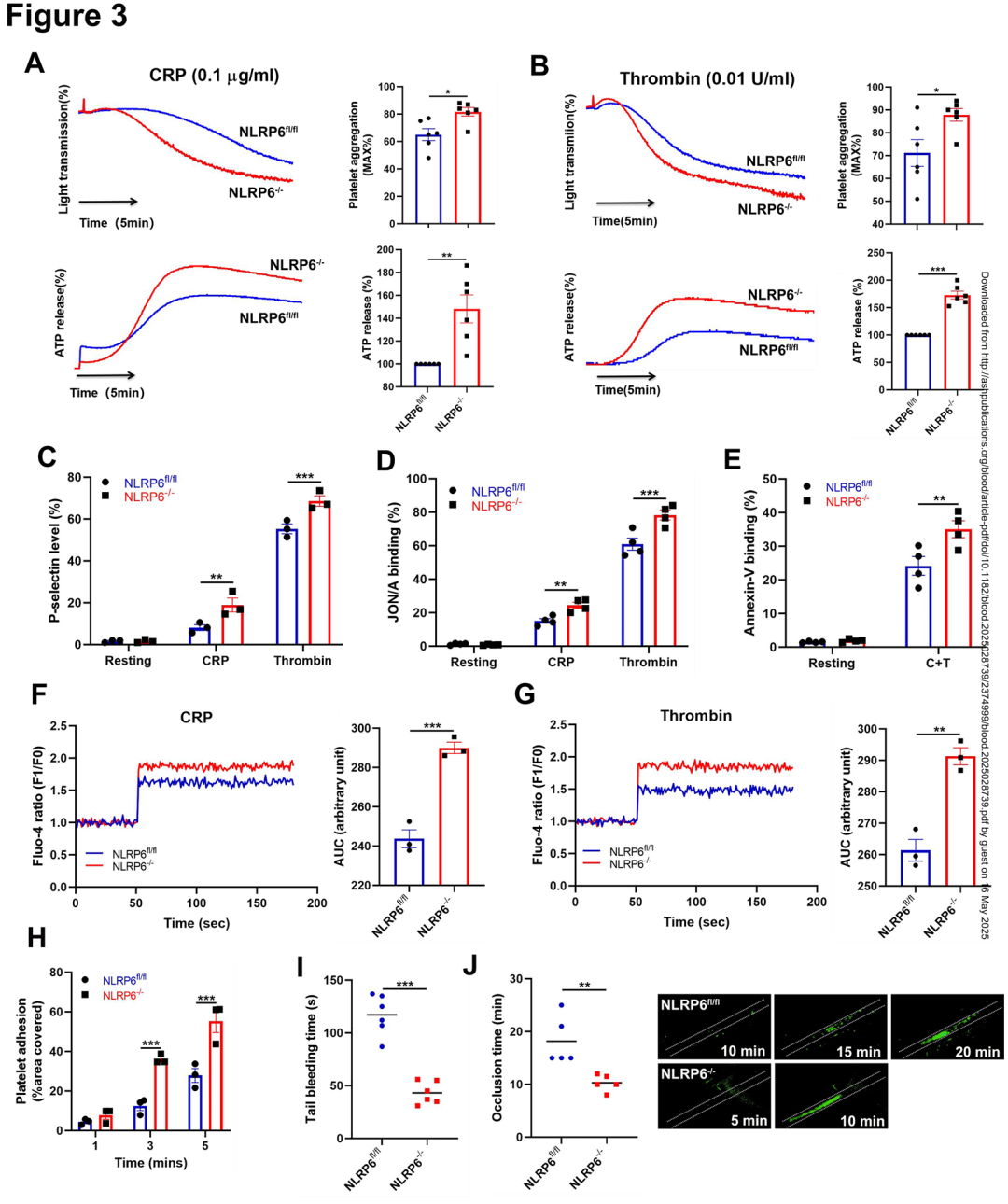
03.NLRP6缺陷导致血小板高反应性和促血栓表型
体外功能实验显示,NLRP6⁻/⁻ 血小板对胶原相关肽(CRP)和凝血酶的反应性显著增强:CRP(0.1 μg/mL)诱导的聚集率提升28%(82% vs. 64%),ATP 释放量(致密颗粒标志)增加35%(3.8 nM vs. 2.8 nM);凝血酶(0.01 U/mL)刺激下,P-选择素表达(α颗粒标志)升高41%(MFI:1,850 vs. 1,310),活化 αIIbβ3(JON/A结合)增加33%(比例:78% vs. 58%)。
钙流动力学显示,NLRP6 缺失使 CRP 诱导的胞内 Ca²⁺ 峰值升高42%(ΔF/F₀:3.2 vs. 2.3)。流式剪切实验(1,800 s⁻¹)中,NLRP6⁻/⁻ 血小板在胶原表面的黏附面积增加1.5倍。体内实验进一步证实,NLRP6⁻/⁻ 小鼠尾静脉出血时间缩短58%(1.2分钟 vs. 2.9分钟),氯化铁诱导的颈动脉血栓闭塞时间加速42%。这些数据表明,NLRP6 通过负向调控血小板活化、分泌及凝血功能,维持败血症中血栓形成的动态平衡。
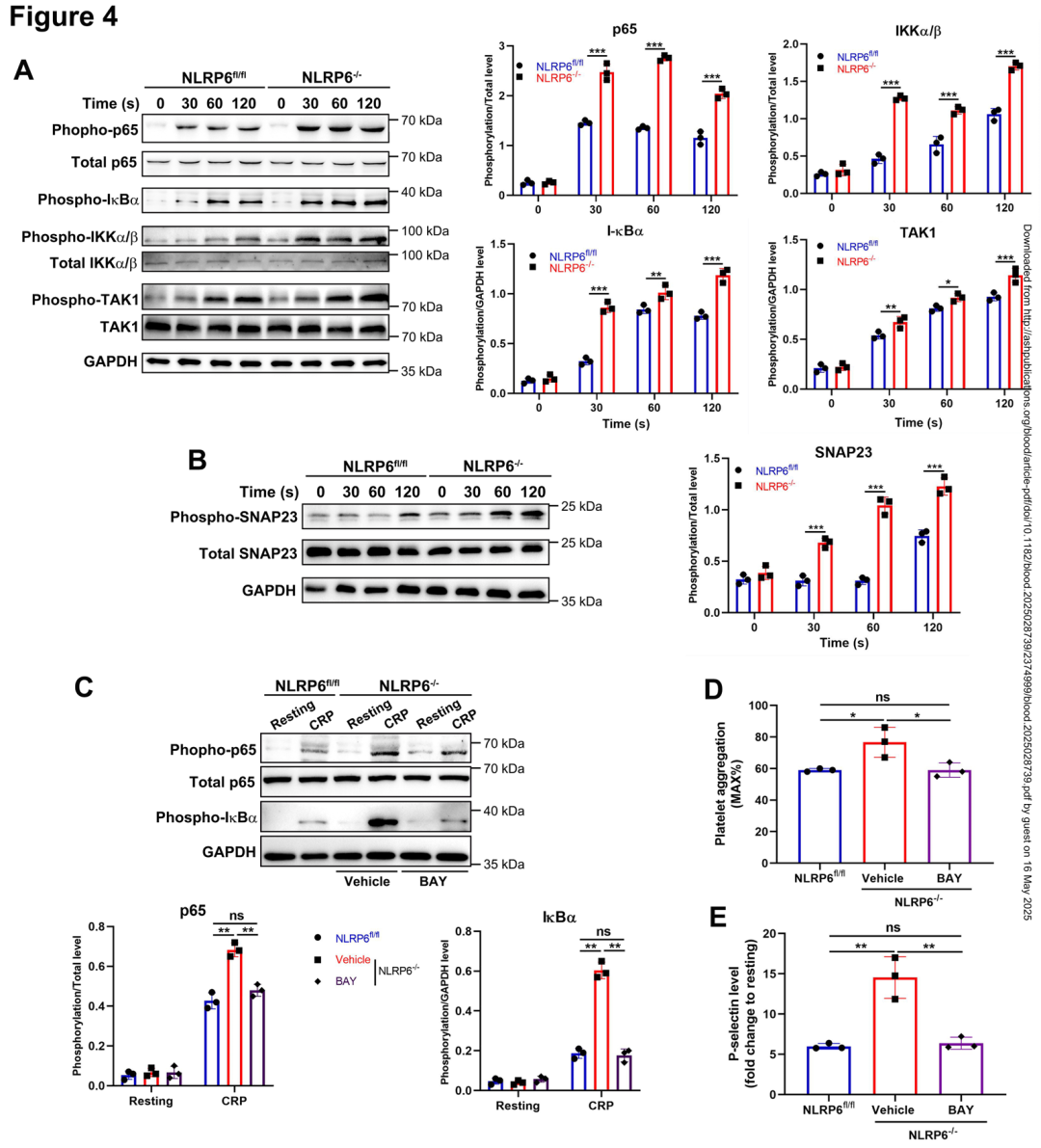
为探索 NLRP6 调控血小板活化的分子机制,作者发现 NLRP6 缺失不改变 IL-1β 或 IL-18 分泌(ELISA检测浓度差异<10%),提示其作用独立于炎症小体。蛋白质印迹显示,CRP 刺激后 NLRP6⁻/⁻ 血小板中 NF-κB 通路关键分子磷酸化水平显著升高:TAK1(Thr184/187)增加2.1倍,IKKα/β(Ser176/180)升高1.9倍,IκBα(Ser32)和p65(Ser536)分别增强2.3倍和2.5倍。SNARE 蛋白 SNAP23(Ser95)磷酸化水平也升高1.7倍(与颗粒分泌相关)。NF-κB 抑制剂 BAY 11-7082(2 μM)预处理可完全逆转 NLRP6⁻/⁻ 血小板的高反应性:CRP诱导的聚集率从82%降至52%,P-选择素表达(MFI)从1,850降至1,120。这些结果首次揭示 NLRP6 通过抑制 NF-κB 信号通路负调控血小板功能。
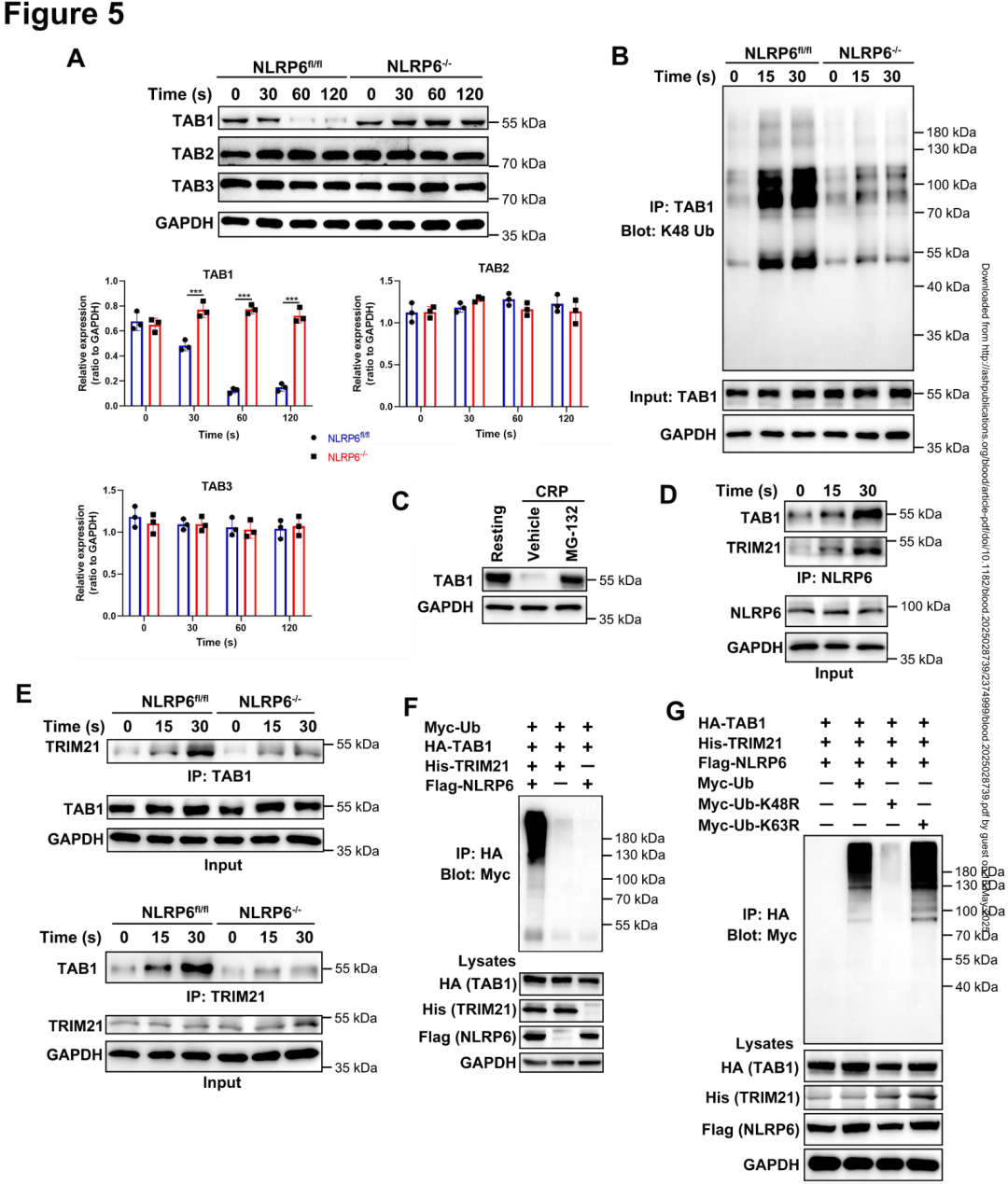
05.NLRP6促进TRIM21与TAB1的相互作用并诱导TAB1降解
机制研究进一步表明,CRP 刺激后野生型血小板中 TAB1 蛋白在60秒内降解80%,而 NLRP6⁻/⁻ 血小板中 TAB1 稳定性显著增强(120秒后残留量仍为60%)。免疫共沉淀(Co-IP)结合质谱分析发现,NLRP6 与 E3 泛素连接酶 TRIM21 及 TAB1 形成复合物。CRP 刺激促进 NLRP6-TRIM21-TAB1 三元复合物的结合(结合强度增加3.5倍),并诱导 TAB1 的 K48 多聚泛素化(泛素化水平升高4.2倍)。蛋白酶体抑制剂 MG-132(20 μM)可完全阻断 TAB1 降解。在 HEK293T 细胞中,共转染 NLRP6、TRIM21 和 TAB1 质粒后,TAB1 的 K48 泛素化水平增加3.8倍,而K63泛素化无变化。突变实验证实,泛素 K48R(而非K63R)完全抑制 TAB1 泛素化。这些数据揭示 NLRP6 作为支架蛋白介导 TRIM21 对 TAB1 的 K48 泛素化修饰,从而通过蛋白酶体途径降解 TAB1,负反馈抑制 NF-κB 信号。
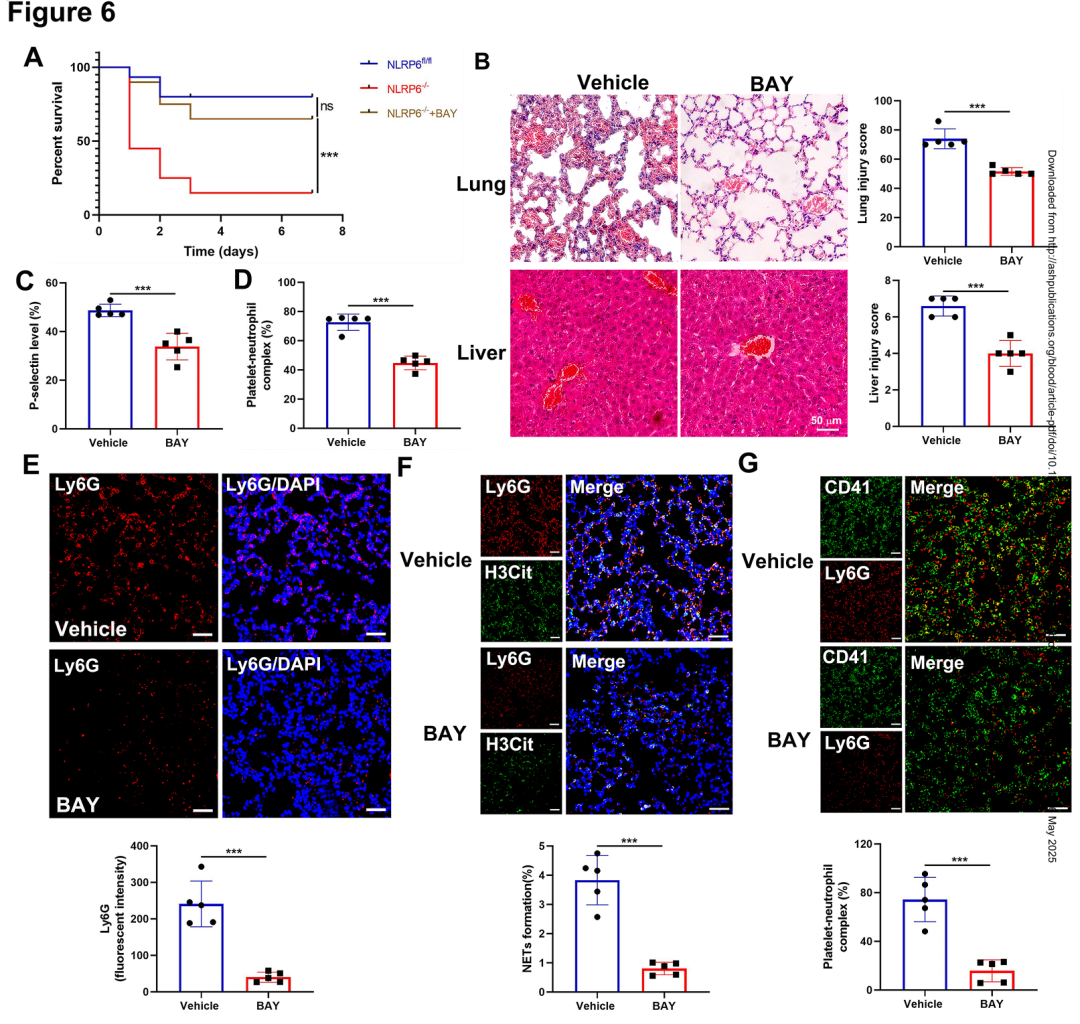
06.抑制NF-kB信号减轻NLRP6缺陷小鼠脓毒症微血栓形成并延长其存活时间
为验证 NF-κB 通路在败血症中的治疗潜力,作者对 NLRP6⁻/⁻ 小鼠术前1小时腹腔注射 BAY 11-7082(10 mg/kg)。结果显示,BAY 处理组72小时存活率从13%提升至47%,肺和肝组织微血栓密度分别降低65%和58%。外周血小板激活(P-选择素)和PNAs比例(15% vs. 34%)均恢复至野生型水平。肺组织免疫荧光显示,中性粒细胞浸润减少1.8倍,NETs 面积缩小58%。这些结果证明,靶向 NF-κB 通路可逆转 NLRP6 缺失导致的败血症恶化,为其临床转化提供依据。
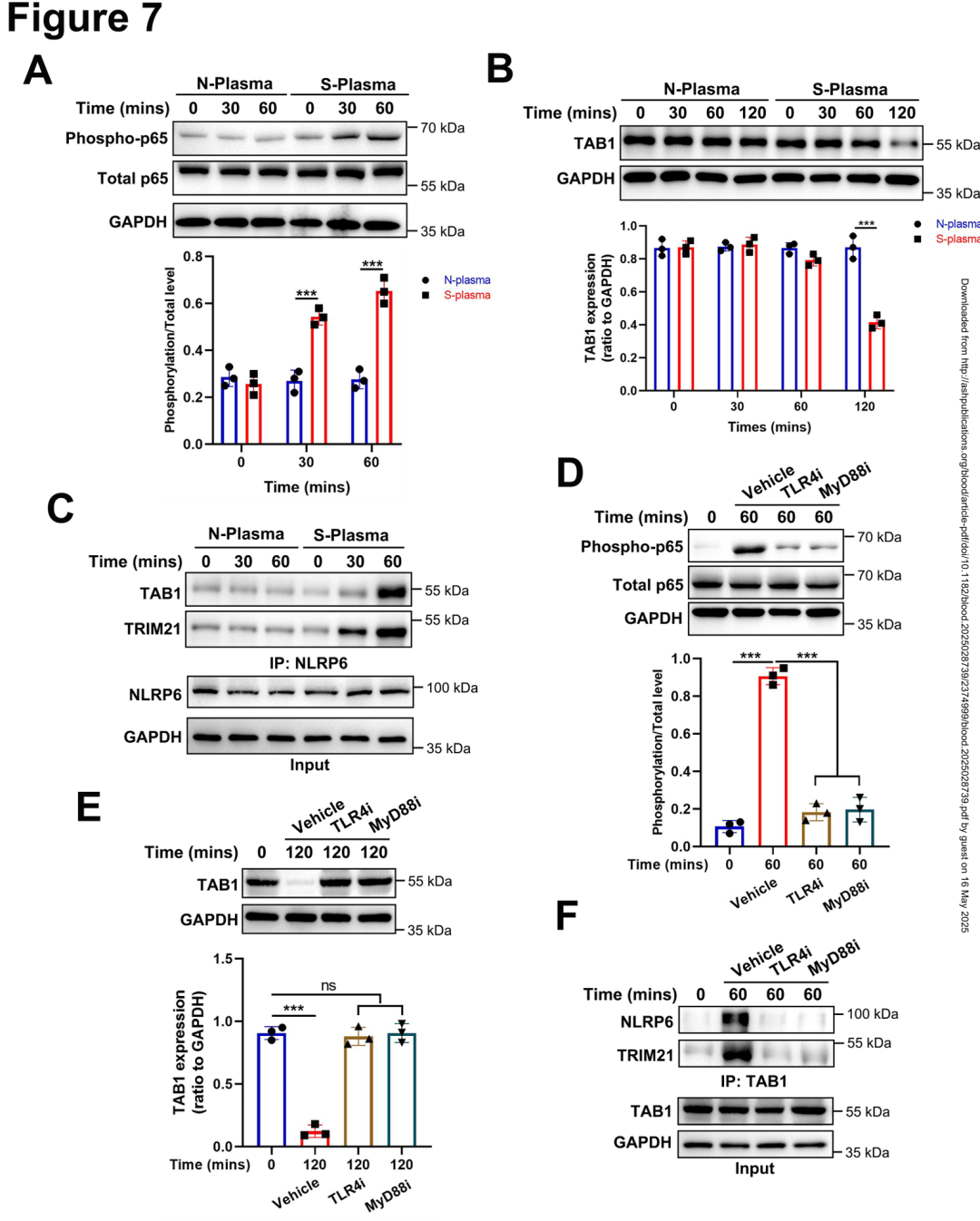
07.脓毒症血浆促进NLRP6与TRIM21和TAB1的相互作用并诱导血小板中TAB1降解
最后,作者探讨了败血症血浆对血小板 NLRP6-TRIM21-TAB1 轴的影响。将野生型血小板与败血症小鼠血浆共孵育,发现 p65 磷酸化水平在30分钟内升高2.3倍,TAB1 蛋白在120分钟后降解82%。Co-IP 实验显示,败血症血浆使 NLRP6-TRIM21 和 NLRP6-TAB1 结合强度分别增加2.8倍和3.2倍。TLR4 抑制剂(TLR4-IN-C34,10 μM)或 MyD88 抑制剂(ST-2825,5 μM)预处理可完全阻断上述效应(p65 磷酸化降低72%,TAB1 降解抑制90%)。这些数据表明,败血症血浆通过 TLR4/MyD88 信号激活血小板 NLRP6-TRIM21 复合体,动态调控 TAB1 降解以限制 NF-κB 过度活化,这一机制在败血症早期发挥关键保护作用。
总之,本研究首次揭示了血小板 NLRP6 通过调控 NF-κB 信号通路在败血症微血管血栓形成中的关键保护作用。实验证实:(1)血小板特异性NLRP6缺失通过增强 TAK1-TAB1 介导的 NF-κB 信号,导致血小板过度活化(聚集率提升28%、α/δ颗粒分泌增加35-40%);(2)异常活化的血小板通过 P-selectin 依赖的相互作用促进中性粒细胞浸润(增加1.9倍)和 NETs 形成(扩大3.7倍),最终加剧肺肝微血栓(面积增加2.3倍)并缩短生存期(72小时死亡率升高67%);(3)分子机制上,NLRP6 作为支架蛋白介导 E3 泛素连接酶 TRIM21 与 TAB1 结合,促进其 K48 泛素化降解(半衰期缩短4倍),从而负调控 NF-κB 信号;(4)败血症血浆通过 TLR4/MyD88 通路激活该调控轴,形成动态负反馈环路(6小时内TAB1降解率达82%)。抑制 NF-κB 信号可完全逆转 NLRP6 缺失的表型(存活率恢复至47%),验证了该通路的中心地位。
该工作首次阐明血小板 NLRP6 在败血症中的保护作用,突破传统认知中NLRP6的促炎角色;此外还揭示了 NLRP6 通过非经典炎症小体途径(不依赖 IL-1β/IL-18 分泌)调控血小板功能,拓展了 NLR 家族蛋白的功能谱;最后,还发现 TRIM21-TAB1-NF-κB 轴在血小板中的全新调控网络,为理解免疫-血栓交互作用提供了分子框架。这些发现不仅解释了败血症患者血小板 NLRP6 表达缺失(临床样本检测显示完全丢失)与微血栓形成的因果关系,更为开发靶向血小板 NLRP6/NF-κB 轴的治疗策略(如 TLR4 抑制剂或 TRIM21 激活剂)奠定了理论基础,具有重要的转化医学价值。