Nature子刊:IL-10靶向IRF转录因子抑制干扰素和炎症
时间 : 2025-04-25
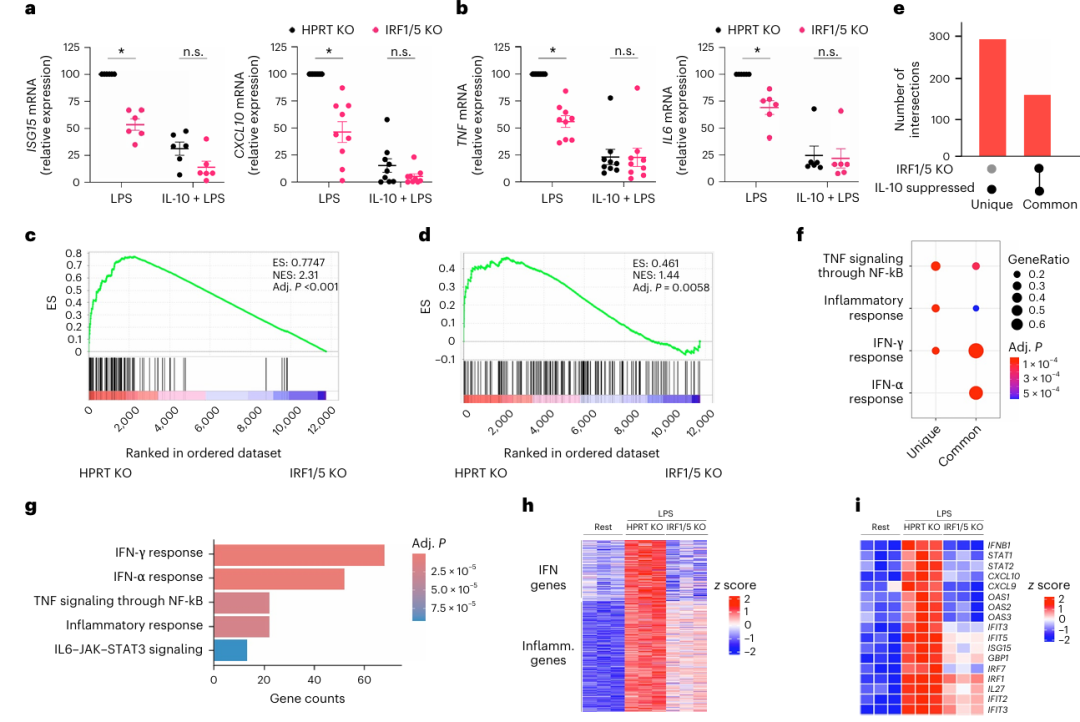
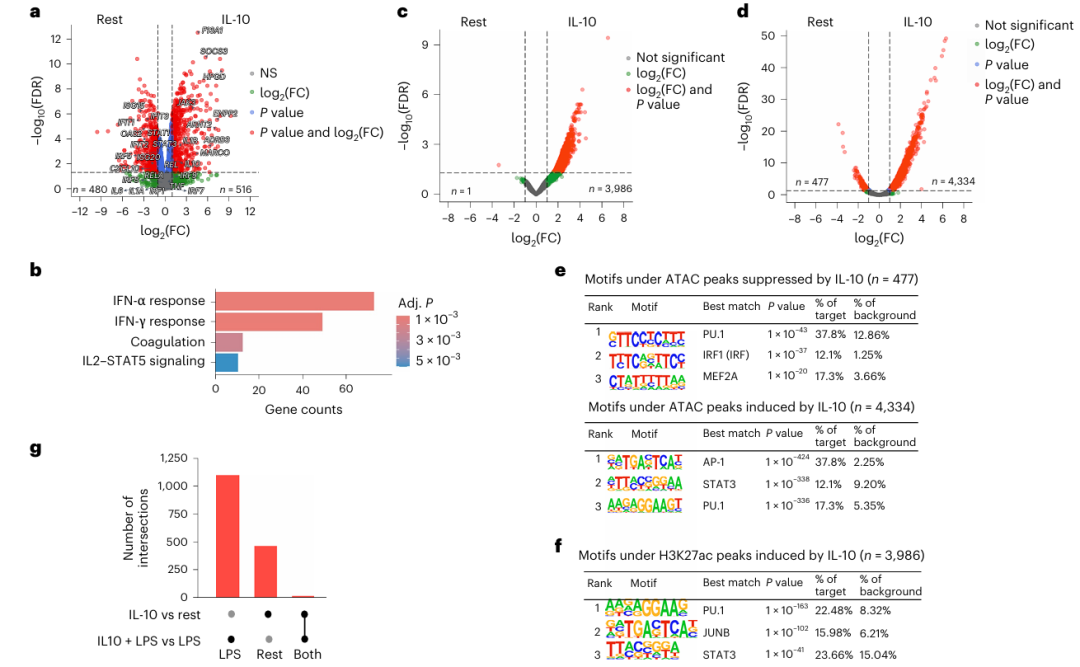
作者首先从健康人外周血中分离了人原代人单核细胞进行实验:将细胞用 IL-10 预处理18小时后,用 LPS 刺激3小时,通过 RT-qPCR 检测基因表达。结果显示,IL-10 显著抑制了 LPS 诱导的经典炎症基因(如IL6、TNF)和干扰素刺激基因(ISGs,如CXCL10、ISG15),且抑制作用在7名独立供体的细胞中一致(Fig a)。进一步通过 RNA-seq 和基因集富集分析发现,IL-10 对 LPS 诱导的干扰素反应的抑制效应相较于炎症反应更显著(Fig b)。聚类分析将 LPS诱导的基因分为了6类,其中被 IL-10 强抑制的基因簇(C2)主要富集于干扰素通路,而中度抑制的基因簇(C3)则富集于 NF-κB 炎症通路(Fig c-d)。核心 IL-10 抑制基因(436个)中,干扰素通路基因的富集程度最高(Fig e),且ISGs(如CXCL10)的抑制程度远高于炎症基因(如IL6)(Fig f-g)。
为验证 IL-10 的广谱性,作者用 TNF 刺激单核细胞6小时,发现 IL-10 同样显著抑制 TNF 诱导的干扰素基因(如CXCL10),但对 TNF 诱导的炎症基因(如TNF本身)抑制较弱(Fig h-j)。此外,IL-10 未抑制 LPS 激活的 NF-κB 信号通路,提示其作用不依赖经典炎症信号阻断。这些结果表明 IL-10 通过靶向干扰素反应基因(而非直接抑制NF-κB或MAPK通路)发挥强效抗炎作用,且这种抑制在干扰素通路中尤为显著。
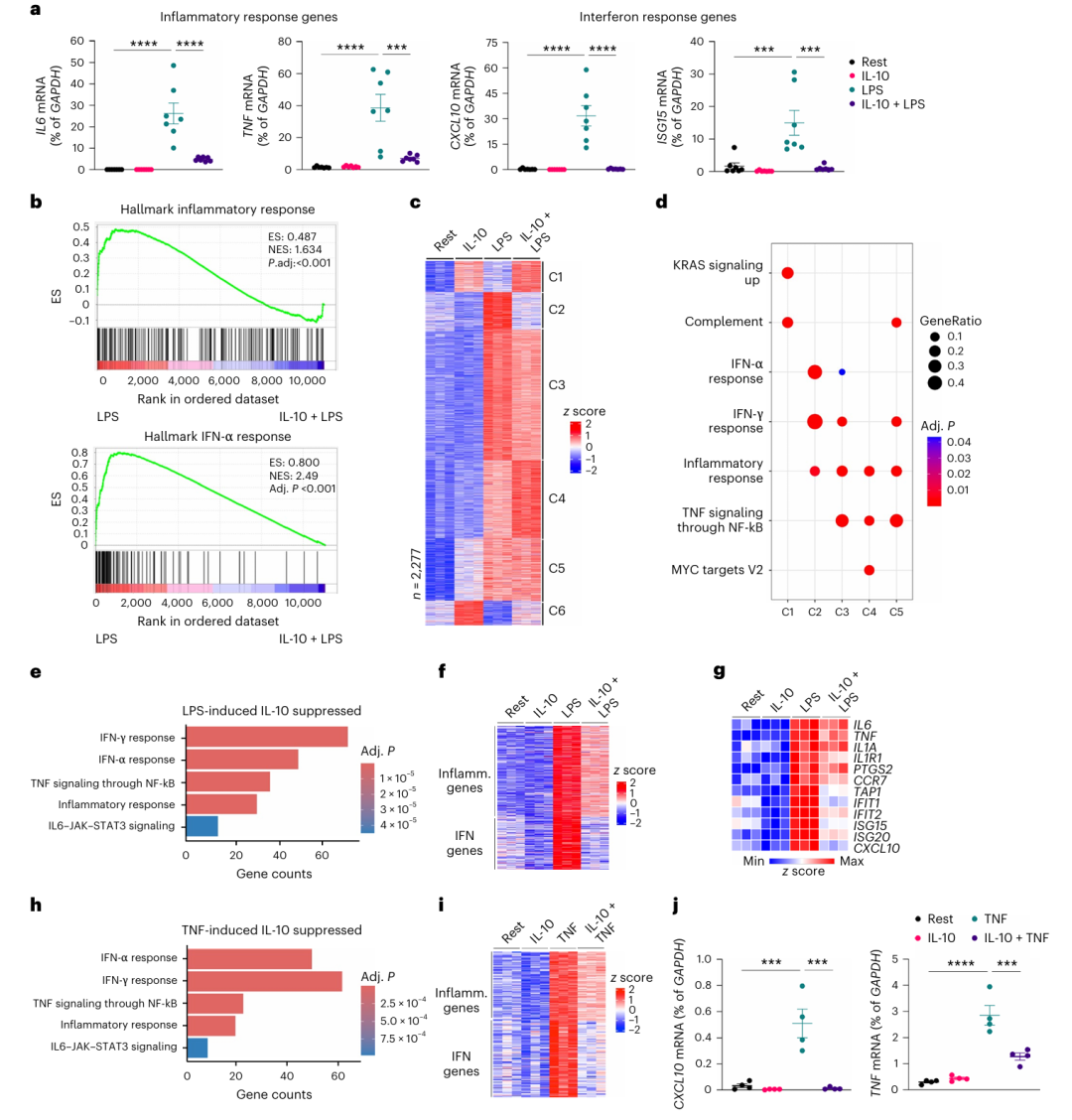
作者进一步利用 CUT&RUN 技术分析组蛋白 H3K27 乙酰化(H3K27ac,活性染色质的标志)在全基因组的变化。结果显示,LPS 诱导的 5749 个H3K27ac峰中,60%(Group 1)被 IL-10 显著抑制至接近基线水平,而剩余 40%(Group 2)仅部分减弱(Fig a-c)。Group 1 峰主要位于基因间区和内含子区(Fig d),且富集 IRF1 结合基序(Fig g);Group 2 峰则主要富集 NF-κB 和 AP-1 基序。通过基因轨迹图(如 IFIT 和 IL6 位点)发现,IL-10 显著降低 Group 1 峰区域的 H3K27ac 和 H3K4me3 (启动子活性标记),而 Group 2 峰区域的 H3K4me3 未受明显影响(Fig e-f),表明 IL-10 对两类基因的调控机制不同。共同说明 IL-10 通过表观遗传机制(如抑制 H3K27ac 和 H3K4me3 修饰)靶向 IRF1 结合的染色质区域,降低其活性,从而优先抑制依赖IRF1的干扰素反应基因(如ISGs),且这种作用独立于 NF-κB 和 AP-1 通路。
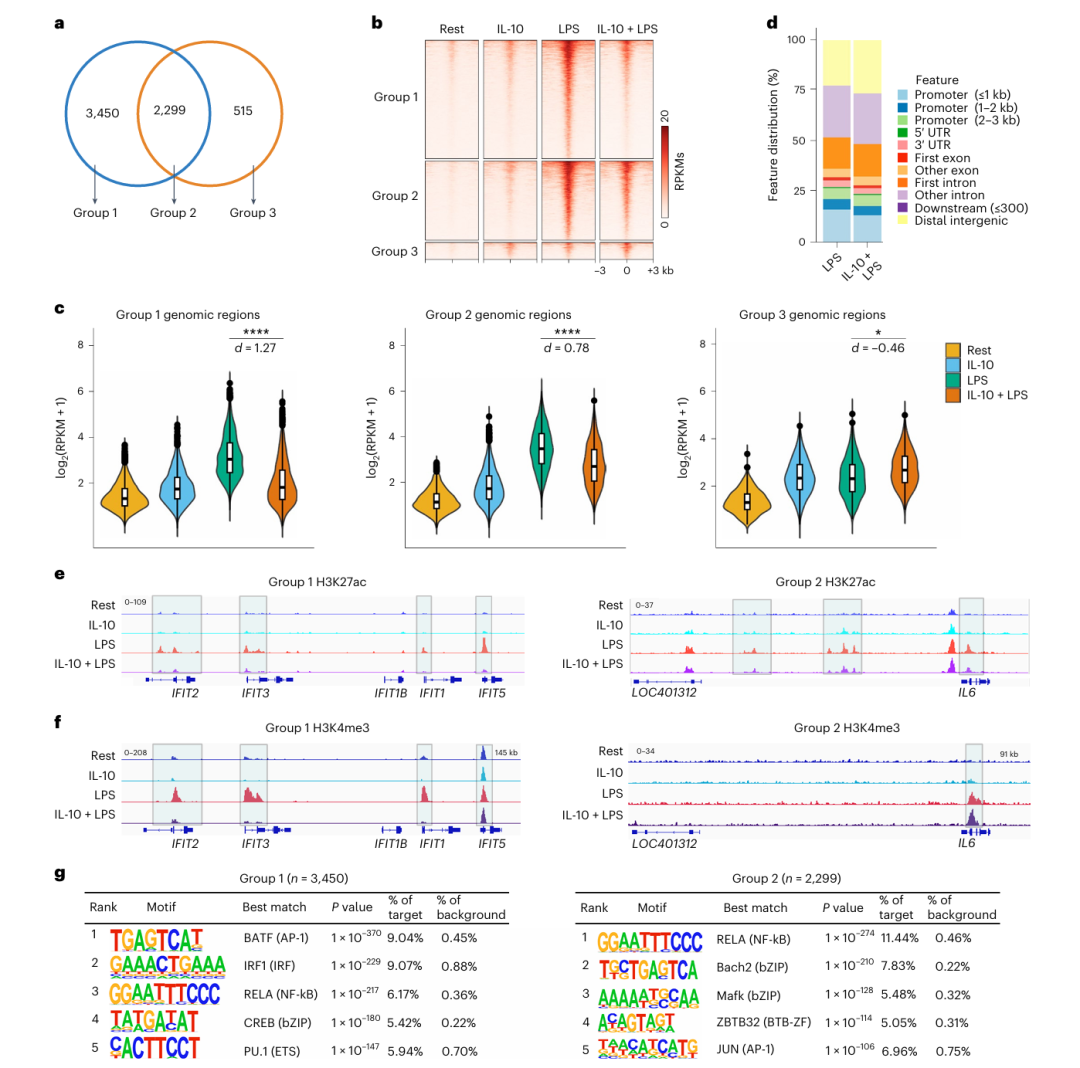
作者通过 ATAC-seq 分析了 IL-10 对 LPS 刺激后人单核细胞染色质可及性的影响。结果显示,LPS 诱导了 8813 个开放染色质区域,其中12.6%(1112个)在 IL-10 预处理后被显著抑制(Fig a-d)。这些被抑制的区域主要位于干扰素刺激基因(ISGs,如IFIT1、ISG15)的基因座,并富集 IRF 和干扰素敏感反应元件(ISRE)的结合基序(Fig e-f)。与此相反,炎症基因(如IL1B)的染色质可及性未受IL-10影响(Fig e),提示IL-10对干扰素通路具有选择性抑制作用。
通过 TOBIAS 和 ChromVAR 计算分析转录因子结合活性发现,IL-10 显著抑制 LPS 诱导的 IRF 家族(如IRF1、IRF5)的 DNA 结合能力和转录活性(Fig h-i),而 NF-κB 和 AP-1 的活性基本保留。类似地,在 TNF 刺激的单核细胞中,IL-10 也选择性抑制 IRF 基序的占据。这些结果表明,IL-10 并非通过阻断经典炎症信号(NF-κB/MAPK),而是通过表观遗传机制抑制 IRF 的染色质结合和激活能力发挥作用。

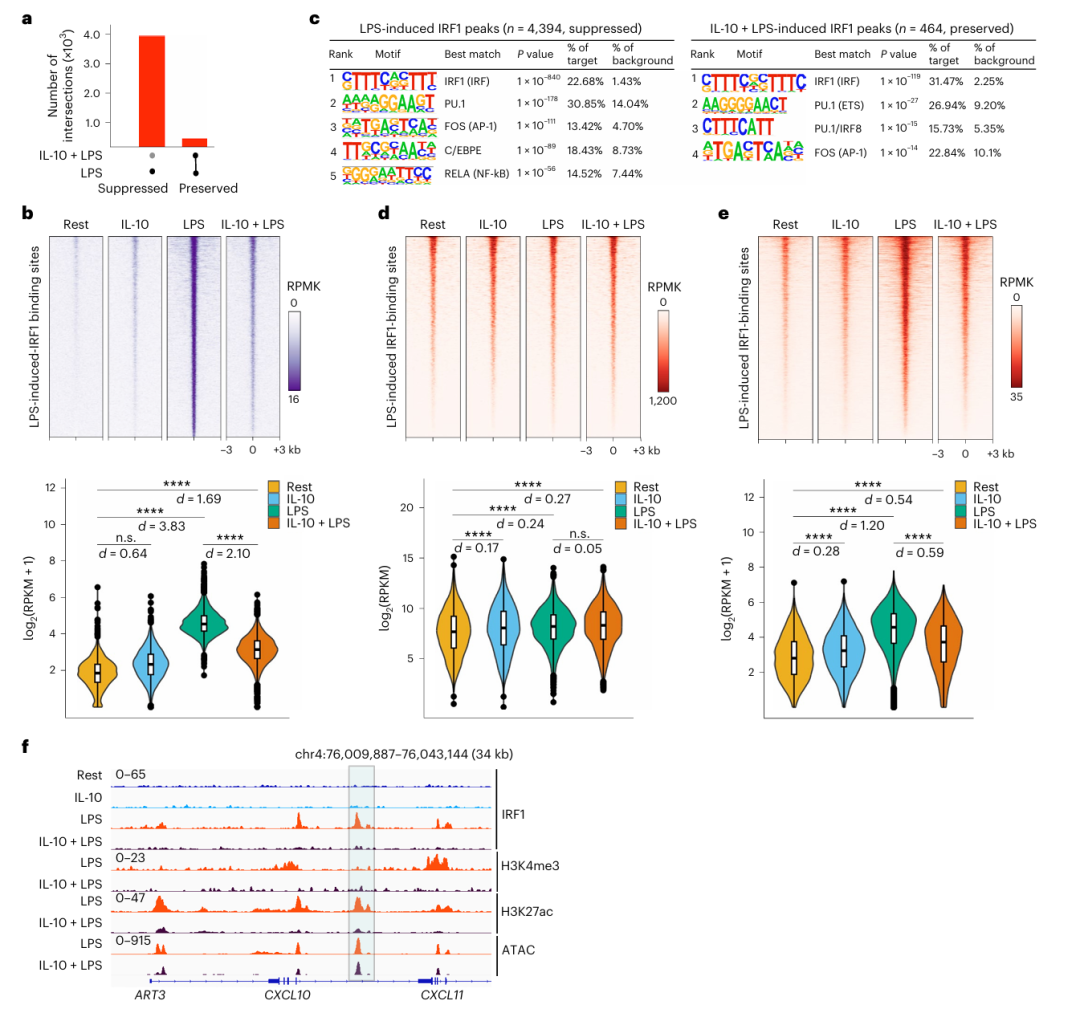
作者通过 CUT&RUN 技术分析了 IL-10 对 LPS 诱导的 IRF1 基因组结合的影响。结果显示,LPS 刺激显著增加了 IRF1 的 DNA 结合位点数量(从基线约200个峰增至4,395个峰),而 IL-10 预处理后,89.5% 的 LPS 诱导 IRF1 结合峰被显著抑制(Fig a-b)。这些被抑制的 IRF1 结合峰主要富集 IRF、AP-1 和 NF-κB 基序,而未被抑制的峰则仅保留 IRF 和 AP-1 基序(Fig c),提示 IL-10 优先靶向依赖 IRF1 与 NF-κB 协同作用的调控区域。
进一步分析染色质状态发现,IRF1 结合的基因组区域在静息单核细胞中已具备开放性(ATAC-seq 信号无显著变化),但 LPS 诱导的 H3K27ac 沉积在 IRF1 结合位点显著增加,而 IL-10 则几乎完全阻断了这一过程(Fig d-e)。例如,CXCL10 基因座在 LPS 刺激后 IRF1 结合增强并伴随 H3K27ac 和 H3K4me3 水平升高,而 IL-10 预处理抑制了这些表观修饰(Fig f)。此外,IL-10 对染色质开放性的影响有限(如 ART3 基因座),表明 IL-10 作用主要通过抑制组蛋白修饰而非改变染色质结构,其通过阻断 IRF1的 DNA 结合能力及相关的 H3K27ac 沉积,抑制增强子和启动子的表观激活,从而下调干扰素和炎症基因表达。
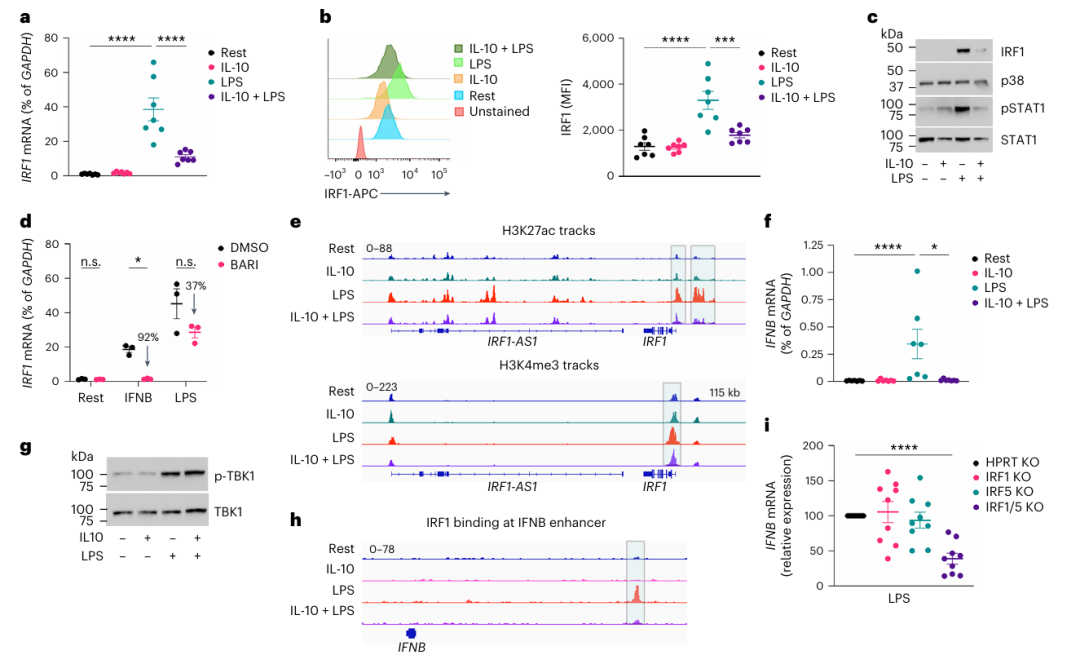
作者通过qPCR和免疫印迹实验发现,IL-10 显著抑制 LPS 诱导的 IRF1 mRNA和蛋白表达(Fig a-c),且这种抑制在 LPS 刺激后2小时即显现。使用 Jak 抑制剂巴瑞替尼(Baricitinib)阻断 IFN-β 自分泌信号后,IRF1 的诱导部分受抑(Fig d),表明 IL-10 通过抑制 IFN-β 自分泌和表观遗传机制(如 STAT3 招募至 IRF1 基因座并降低其 H3K27ac 和 H3K4me3 水平)双重途径抑制 IRF1(Fig e)。
进一步研究发现,IL-10 显著抑制 LPS 诱导的 IFN-β 生成(Fig f),但对 TBK1-IRF3 信号通路的激活无影响(Fig g)。通过 CRISPR-Cas9 敲除 IRF1 或 IRF5 基因发现,单独敲除 IRF1 不影响 LPS 诱导的 IFN-β,但联合敲除IRF1 和 IRF5 (IRF1/5-KO)使 IFN-β 表达下降61%(Fig i),表明 IRF1 与 IRF5 协同驱动 LPS 诱导的 IFN-β 生成。