

Science:肝脏ALKBH5通过双通路调控糖脂代谢
时间 : 2025-03-18哈尔滨工业大学团队于2025年2月28日在《Science》上发表文章 Liver ALKBH5 regulates glucose and lipid homeostasis independently through GCGR and mTORC1 signaling

维持葡萄糖和脂质稳态对健康至关重要,其失调会导致代谢疾病,如2型糖尿病(T2DM)和代谢功能障碍相关脂肪性肝病(metabolic dysfunction–associated fatty liver disease ,MAFLD)。本研究鉴定出RNA N6-甲基腺苷去甲基酶ALKBH5(烷基化修复同源蛋白5)为代谢疾病的主要调控因子。ALKBH5在肥胖期间肝脏中表达上调,并被蛋白激酶A磷酸化,导致其转运至细胞质。肝细胞特异性缺失Alkbh5通过抑制胰高血糖素受体( glucagon receptor,GCGR)和哺乳动物雷帕霉素靶蛋白复合物1( rapamycin complex 1 ,mTORC1)信号通路,降低葡萄糖和脂质水平。靶向敲低肝脏Alkbh5可逆转糖尿病小鼠的T2DM和MAFLD,凸显其治疗潜力。本研究揭示了一种调控机制,即ALKBH5通过整合GCGR和mTORC1通路协调葡萄糖和脂质稳态,为代谢疾病的调控提供了新的见解。
RNA结合蛋白(RNA-binding proteins ,RBPs)是一类多样化的蛋白质,能够与细胞中的双链和单链RNA相互作用,在RNA加工活动中发挥关键作用,包括RNA加帽、多聚腺苷酸化、修饰、剪接、稳定、定位和翻译。一些RBPs对维持生命过程至关重要。越来越多的证据表明,某些RBPs能够响应代谢信号和刺激调控葡萄糖或脂质稳态,其失调会导致代谢疾病的发生。然而,目前尚不清楚RBPs是否能够整合调控葡萄糖和脂质稳态。研究人员假设,特定的代谢相关RBPs可能协调调控肝脏葡萄糖和脂质稳态。
01
一、ALKBH5通过Ser362磷酸化调控肝脏葡萄糖生成在代谢疾病中的作用

db/db BKS小鼠是代谢疾病的常用模型。通过定量蛋白质组学分析,发现db/db小鼠肝脏中有137种蛋白质上调,包括7种RBPs,其中ALKBH5是一种关键的RNA m6A去甲基酶。ALKBH5在db/db小鼠、高脂饮食诱导的肥胖小鼠及人类糖尿病肝脏中表达增加,并通过胰高血糖素-PKA途径在Ser362位点发生磷酸化。结构分析表明,Ser362易于被PKA识别,而Ser253则不易被磷酸化。肝细胞特异性Alkbh5敲除小鼠显示血糖降低、葡萄糖耐量增强及肝脏葡萄糖生成(hepatic glucose production,HGP)减少,这些变化与胰岛素敏感性无关。S362A突变小鼠进一步验证了Ser362磷酸化的重要性,其空腹血糖和葡萄糖耐量显著改善,且对高脂饮食诱导的高血糖具有保护作用。ALKBH5通过Ser362磷酸化调控HGP,其缺失或突变可改善葡萄糖稳态,为代谢疾病治疗提供了潜在靶点。
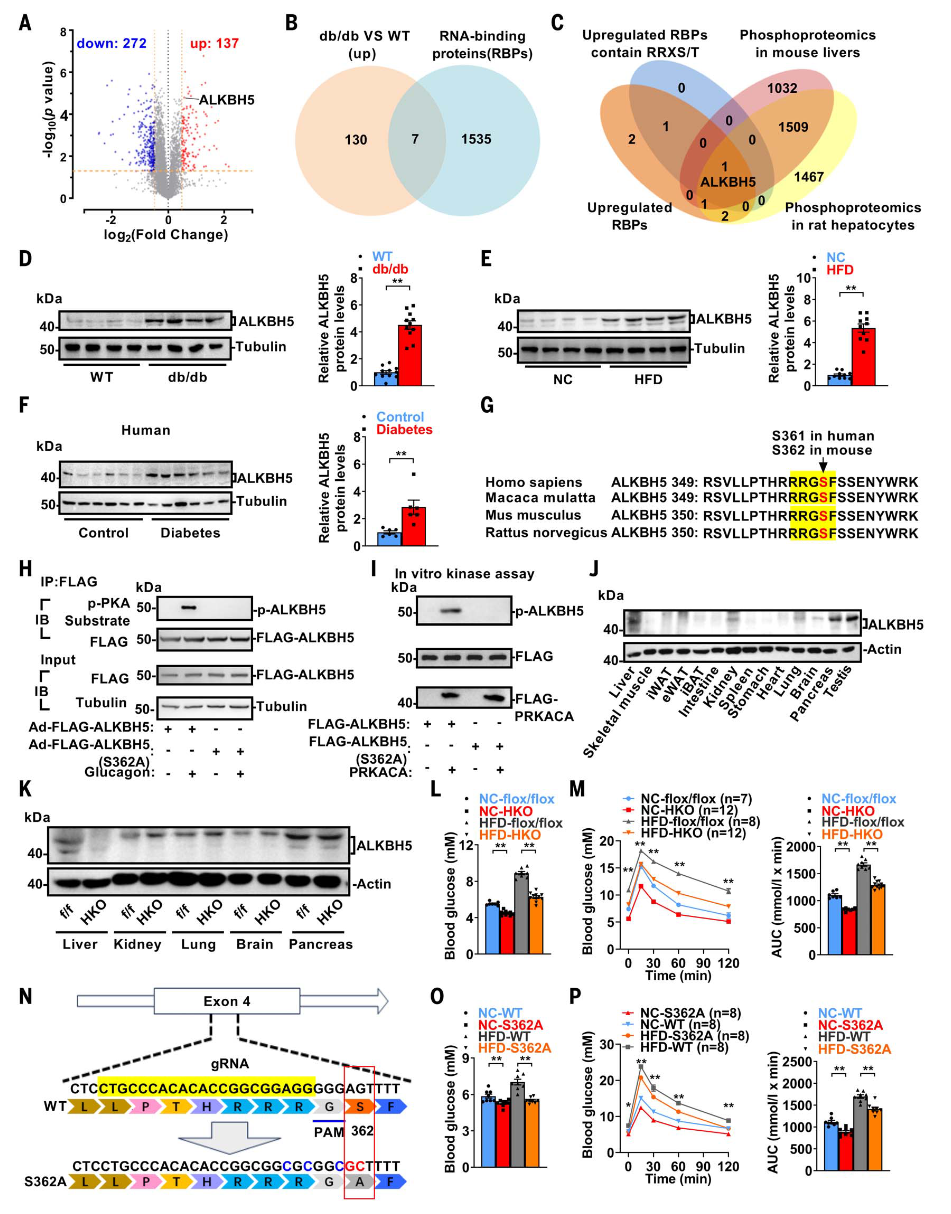
图1.定量蛋白质组学分析鉴定ALKBH5为代谢疾病的关键调控因子
02
二、ALKBH5通过GCGR调控葡萄糖稳态
体内实验表明,肝脏Alkbh5缺失或S362A突变通过降低胰高血糖素敏感性改善葡萄糖稳态。进一步分析聚焦于胰高血糖素-GCGR-cAMP-PKA-CREB信号通路。尽管Alkbh5-HKO小鼠血清胰高血糖素水平与对照组相似,但其肝脏GCGR、cAMP、磷酸化PKA底物和磷酸化CREB水平均降低,且糖异生相关基因表达下调。S362A突变小鼠也表现出类似的GCGR-cAMP-PKA-CREB信号通路减弱和糖异生基因表达下降。这些结果表明,ALKBH5及其Ser362磷酸化在维持GCGR-cAMP-CREB信号通路中起关键作用。
通过恢复GCGR表达,Alkbh5-HKO小鼠的血糖水平升高,葡萄糖耐量受损,肝脏葡萄糖生成增加,胰高血糖素敏感性增强,且GCGR-cAMP-PKA-CREB信号通路被重新激活。此外,使用腺苷酸环化酶激活剂forskolin可逆转Alkbh5-HKO肝细胞中GCGR-PKA-CREB信号的下调,而过表达ALKBH5则增加GCGR蛋白水平并激活PKA信号,这一效应可被PKA抑制剂H89抑制。这些发现表明,ALKBH5通过GCGR和PKA信号通路调控葡萄糖稳态。
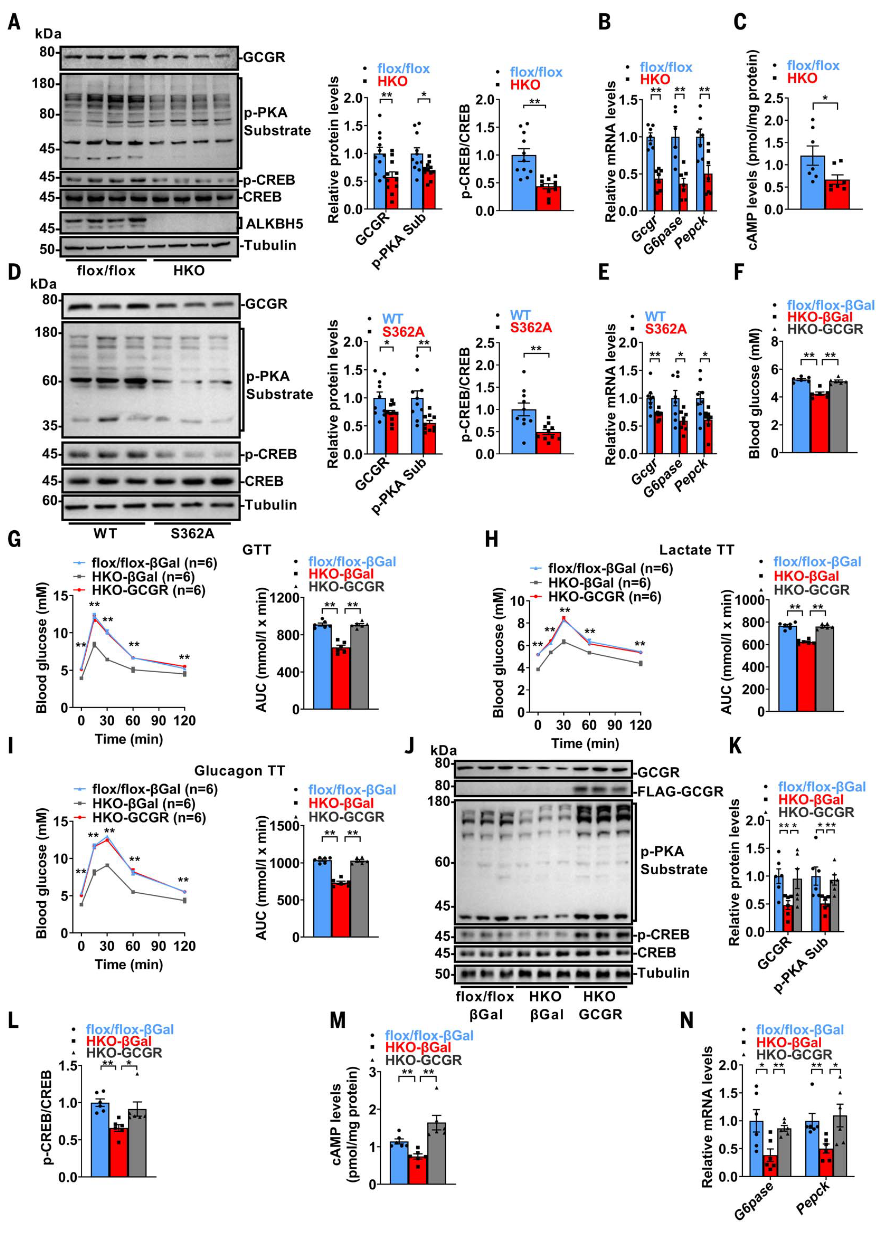
图2.ALKBH5通过GCGR调控葡萄糖稳态
03
三、ALKBH5通过其去甲基化活性促进Gcgr mRNA的稳定
ALKBH5是一种m6A去甲基酶,其缺失导致肝脏m6A水平升高。通过m6A-RNA免疫沉淀测序分析,发现Alkbh5-HKO小鼠肝脏中2339个转录本的m6A修饰增加,1766个转录本的m6A修饰减少。RNA测序显示,Alkbh5-HKO小鼠肝脏中1118个基因表达上调,968个基因表达下调。进一步整合MeRIP-seq和RNA-seq数据发现,190个m6A修饰增加的转录本与mRNA表达水平下降相关,其中包括与胰高血糖素信号和糖异生相关的Gcgr基因。Gcgr mRNA的m6A修饰增加导致其表达水平下降,表明ALKBH5可能通过其去甲基化活性增强Gcgr表达。
为验证这一假设,研究人员构建了去甲基化活性缺失的ALKBH5突变体(H205A),并通过腺病毒或腺相关病毒进行功能实验。结果显示,ALKBH5(而非H205A突变体)能够恢复Alkbh5-HKO小鼠肝脏中GCGR及其下游信号通路的表达,并改善葡萄糖稳态相关表型,包括血糖水平、葡萄糖耐量、乳酸诱导的葡萄糖生成和胰高血糖素敏感性。此外,ALKBH5通过其去甲基化活性增强了Gcgr mRNA的稳定性,而H205A突变体则无此作用。
进一步研究发现,ALKBH5直接与Gcgr转录本的m6A修饰区域结合,并通过m6A修饰调控Gcgr mRNA的稳定性。YTHDF2和YTHDF3(而非YTHDF1)能够识别Gcgr转录本的m6A修饰并降低其稳定性。此外,Mettl3或Wtap的肝细胞特异性缺失虽然减少了Gcgr转录本的m6A修饰,但由于YTHDF2和YTHDF3蛋白水平升高,并未导致GCGR表达增加或其下游信号通路激活。FTO作为另一种m6A去甲基酶,未显示对Gcgr mRNA或蛋白水平的调控作用。
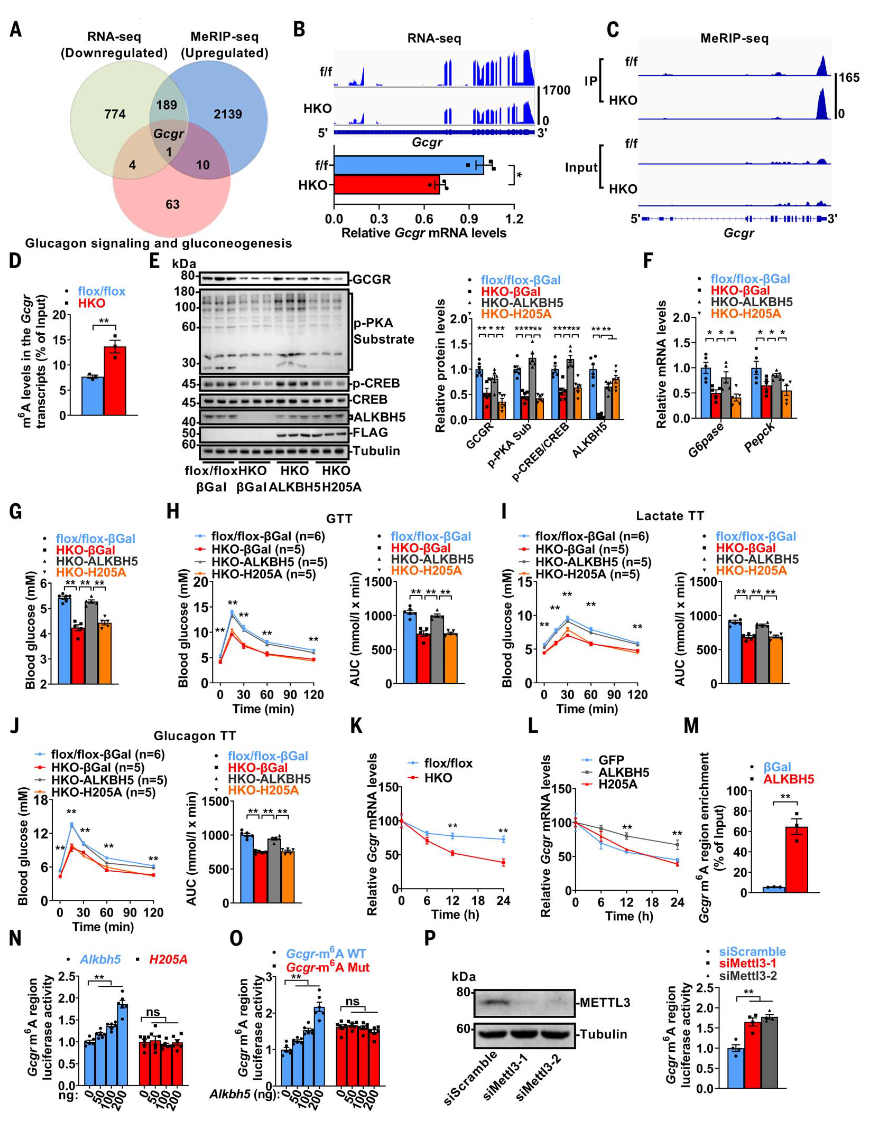
图3.3ALKBH5通过其去甲基化活性增强Gcgr mRNA的稳定性
04
四、 肝细胞特异性缺失Alkbh5预防高脂饮食诱导的MAFLD和高脂血症
在高脂饮食条件下,肝细胞特异性缺失Alkbh5的小鼠表现出对MAFLD和高脂血症的保护作用,表现为肝脏甘油三酯水平降低、脂滴减少、血清肝脏甘油三酯和总胆固醇水平下降、血清丙氨酸氨基转移酶活性降低以及慢性炎症因子表达减少。Alkbh5-HKO小鼠的总体重和白色脂肪组织重量未受影响,且其中的脂解相关信号通路也未改变。肝脏中新生脂质合成速率降低,但β-氧化未受影响,提示保护作用可能与新生脂质合成减少有关。
进一步研究发现,Alkbh5-HKO小鼠肝脏中SREBP1的成熟形式下调,导致关键脂质合成酶及游离脂肪酸转运蛋白CD36的表达降低。mTORC1和AMPK是SREBP1的关键调控因子,Alkbh5-HKO小鼠中p-AMPK水平未变,但p-mTOR及其下游靶点p-S6K水平下降,p-ULK1水平也降低,促进了自噬的激活。PI3K活性在Alkbh5-HKO小鼠肝脏中降低,伴随p-p55和p-AKT水平下降,而INSR水平未变。RNA-seq分析显示,Alkbh5-HKO小鼠肝脏中EGFR和Epha2表达下调,导致EGFR-PI3K-AKT-mTORC1信号通路减弱。这些数据表明,EGFR-PI3K-AKT-mTORC1信号通路的减弱在Alkbh5-HKO小鼠脂质稳态改善中起主要作用。
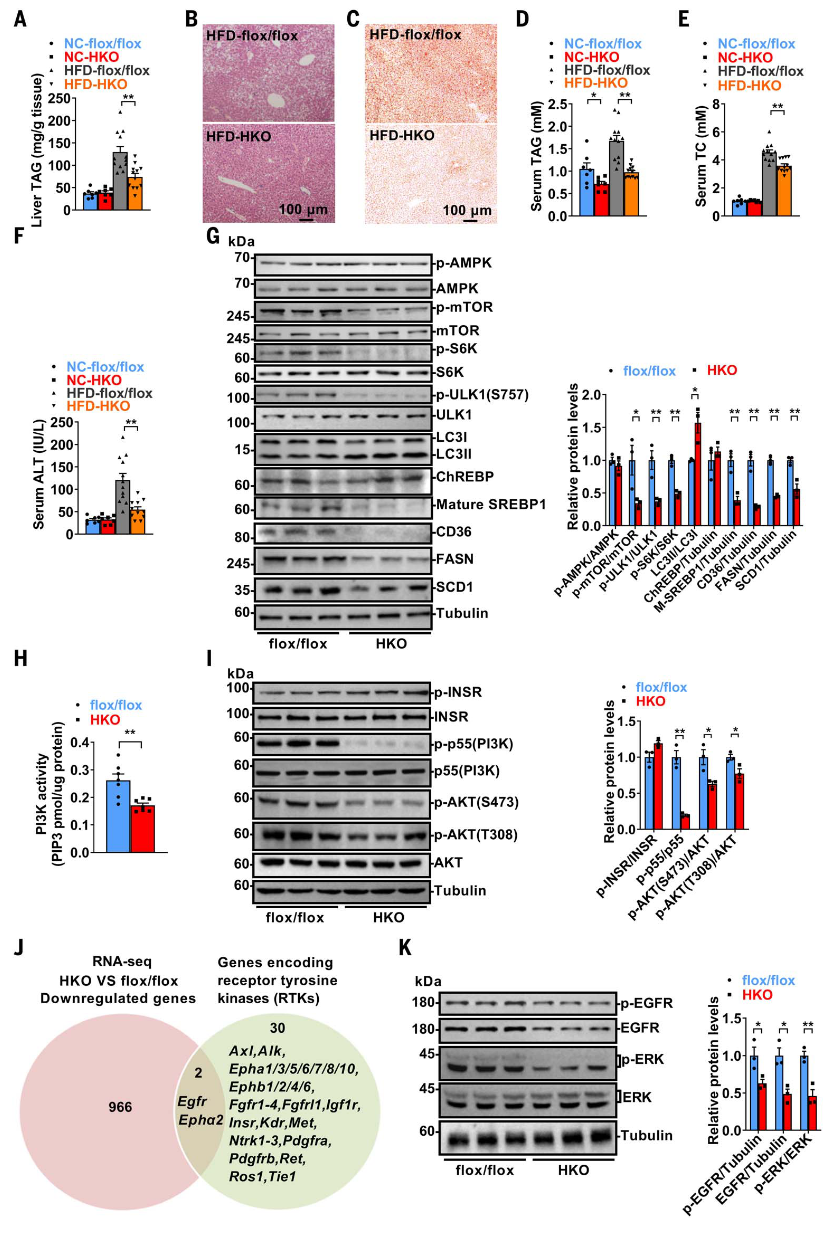
图4.肝细胞特异性缺失Alkbh5预防高脂饮食诱导的脂肪变性和高脂血症
05
五、ALKBH5通过EGFR-PI3K-AKT-mTORC1信号通路调控MAFLD
为研究EGFR是否介导ALKBH5在脂质稳态中的功能,研究人员进行了EGFR恢复实验。在Alkbh5-HKO小鼠分离的原代肝细胞中,通过Ad-EGFR腺病毒恢复EGFR表达,逆转了p-EGFR、p-PI3K p55、p-AKT、p-mTOR、p-ULK1、p-S6K、成熟(切割)SREBP1、CD36、FASN和SCD1水平的下降。同样,在Alkbh5-HKO小鼠肝脏中恢复EGFR表达,逆转了这些信号分子的减少和LC3II/LC3I比率的增加,导致肝脏TAG、血清TAG、血清TC水平和血清ALT活性升高。ALKBH5过表达也提高了EGFR水平,随后增加了EGFR-PI3K-AKT-mTORC1信号通路组分的磷酸化以及成熟SREBP1、CD36、FASN和SCD1的量。EGFR抑制剂拉帕替尼有效阻断了ALKBH5诱导的这些信号通路的激活。这些结果表明,ALKBH5通过EGFR-PI3K-AKT-mTORC1信号通路影响MAFLD。
胰高血糖素信号通路的激活抑制胰岛素诱导的mTORC1和SREBP1通路激活,从而预防MAFLD(31, 32),提示胰高血糖素通路激动剂可能治疗MAFLD。然而,在Alkbh5-HKO小鼠肝脏中,GCGR表达和胰高血糖素信号的减少并未增加mTORC1或成熟(切割)SREBP1信号,也未导致MAFLD。相反,Alkbh5-HKO小鼠表现出mTORC1-成熟SREBP1信号减弱和MAFLD减轻,表明GCGR和mTORC1通路在此背景下独立调控。恢复GCGR不影响肝脏TAG、血清TAG或TC,恢复EGFR-mTORC1也不改变Alkbh5-HKO小鼠的血糖
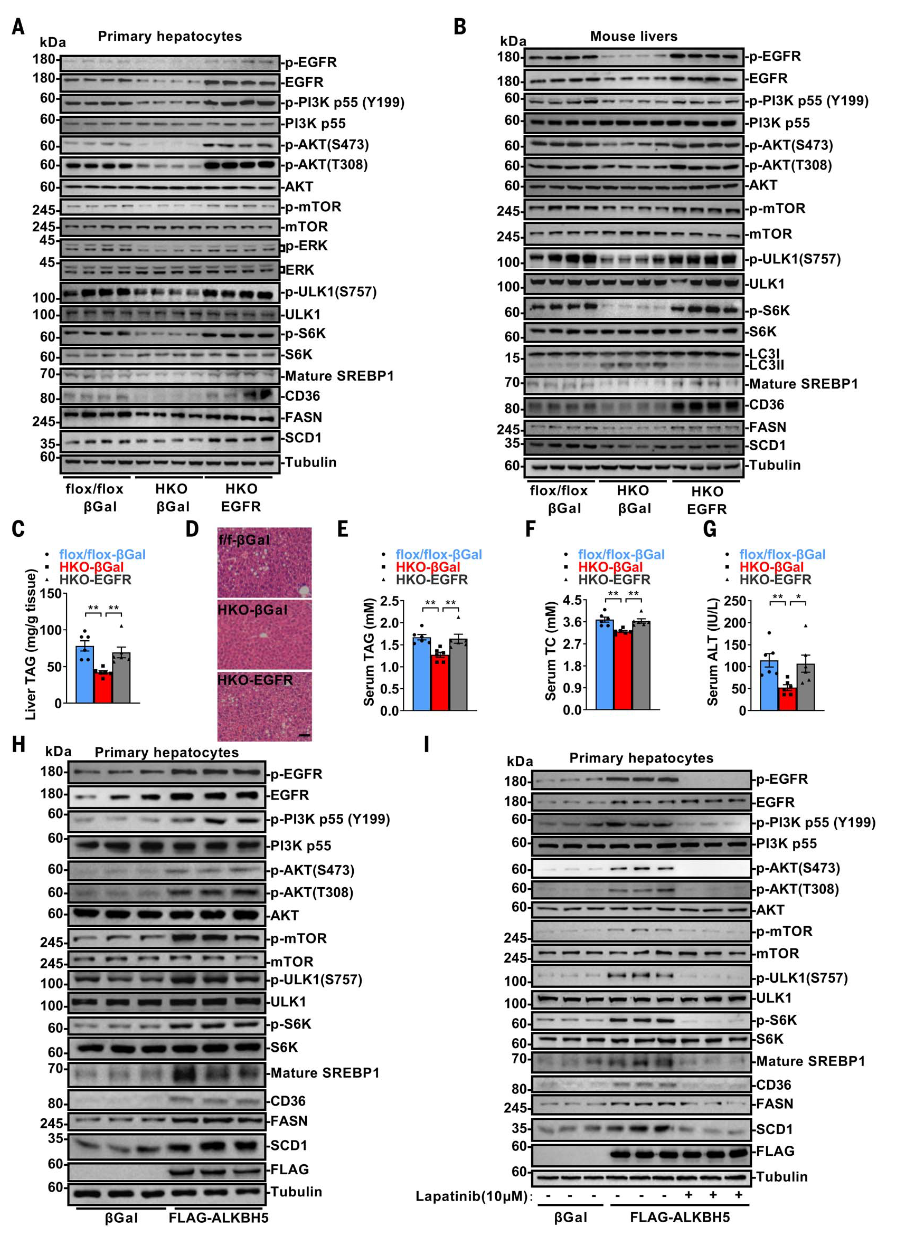
图5.ALKBH5通过EGFR-PI3K-AKT-mTORC1信号通路调控MAFLD
06
六、ALKBH5通过结合Egfr内含子增强子促进EGFR-mTORC1信号通路
ALKBH5及其去甲基化活性缺失突变体均能增加EGFR及其下游信号分子(如p-EGFR、p-AKT、p-mTOR等)的水平,促进肝脏TAG积累及血清TAG和TC升高。ALKBH5通过结合Egfr基因内含子增强子调控其转录,这一过程不依赖于其去甲基化活性。CUT&Tag和CUT&RUN分析显示,ALKBH5结合富含GC的区域,并鉴定出8937个与峰值相关的基因,涉及mTOR信号、自噬等通路。实验证实,ALKBH5直接结合Egfr内含子增强子,并通过其Gln145-Gly152和Cys231-Glu242环与DNA螺旋相互作用,增强Egfr转录。缺失这两个环的ALKBH5突变体无法结合增强子或激活EGFR-mTORC1信号通路。这些结果表明,ALKBH5通过其特定环结构结合Egfr增强子,促进EGFR-mTORC1信号通路,调控脂质稳态。
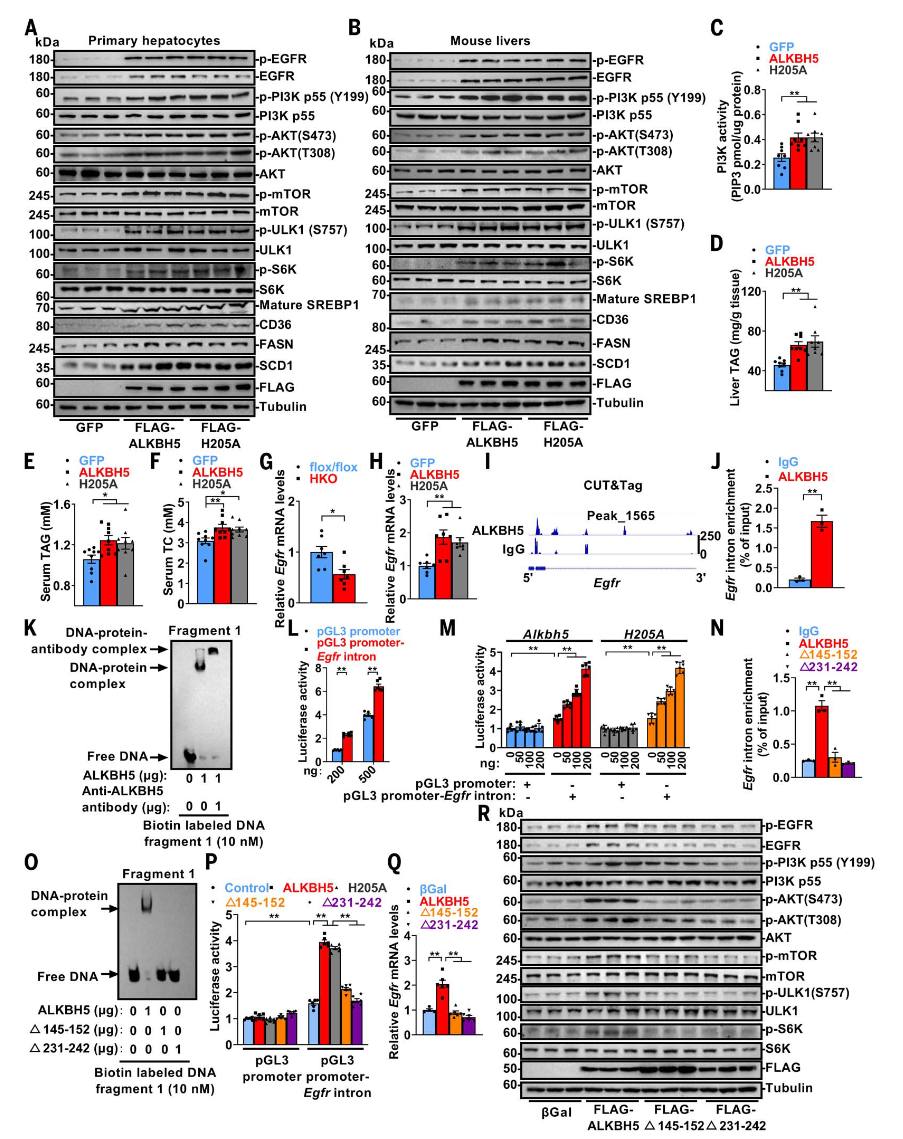
图6.ALKBH5通过增加Egfr转录促进EGFR-PI3K-AKT-mTORC1信号通路,不依赖其去甲基化活性
07
七、Gln145-Gly152和Cys231-Glu242环对ALKBH5调控脂质稳态至关重要,但对葡萄糖稳态非必需
为研究Gln145-Gly152和Cys231-Glu242环在ALKBH5通过EGFR-mTORC1信号通路调控脂质稳态中的作用,研究人员在Alkbh5-HKO小鼠中恢复了ALKBH5、∆Gln145-Gly152或∆Cys231-Glu242的表达。结果显示,∆Gln145-Gly152和∆Cys231-Glu242突变体无法逆转肝脏TAG、血清TAG、TC和ALT活性的降低,而WT ALKBH5成功恢复了这些指标。此外,这两种缺失突变体也无法恢复EGFR及其下游EGFR-PI3K-AKT-mTORC1信号通路的激活,而WT ALKBH5则能有效恢复。然而,ALKBH5及其两种缺失突变体均能恢复Alkbh5-HKO小鼠的血糖水平和GCGR信号通路。这些数据表明,Gln145-Gly152和Cys231-Glu242环对ALKBH5通过EGFR-mTORC1信号通路调控脂质稳态至关重要,但对葡萄糖稳态的调控并非必需。
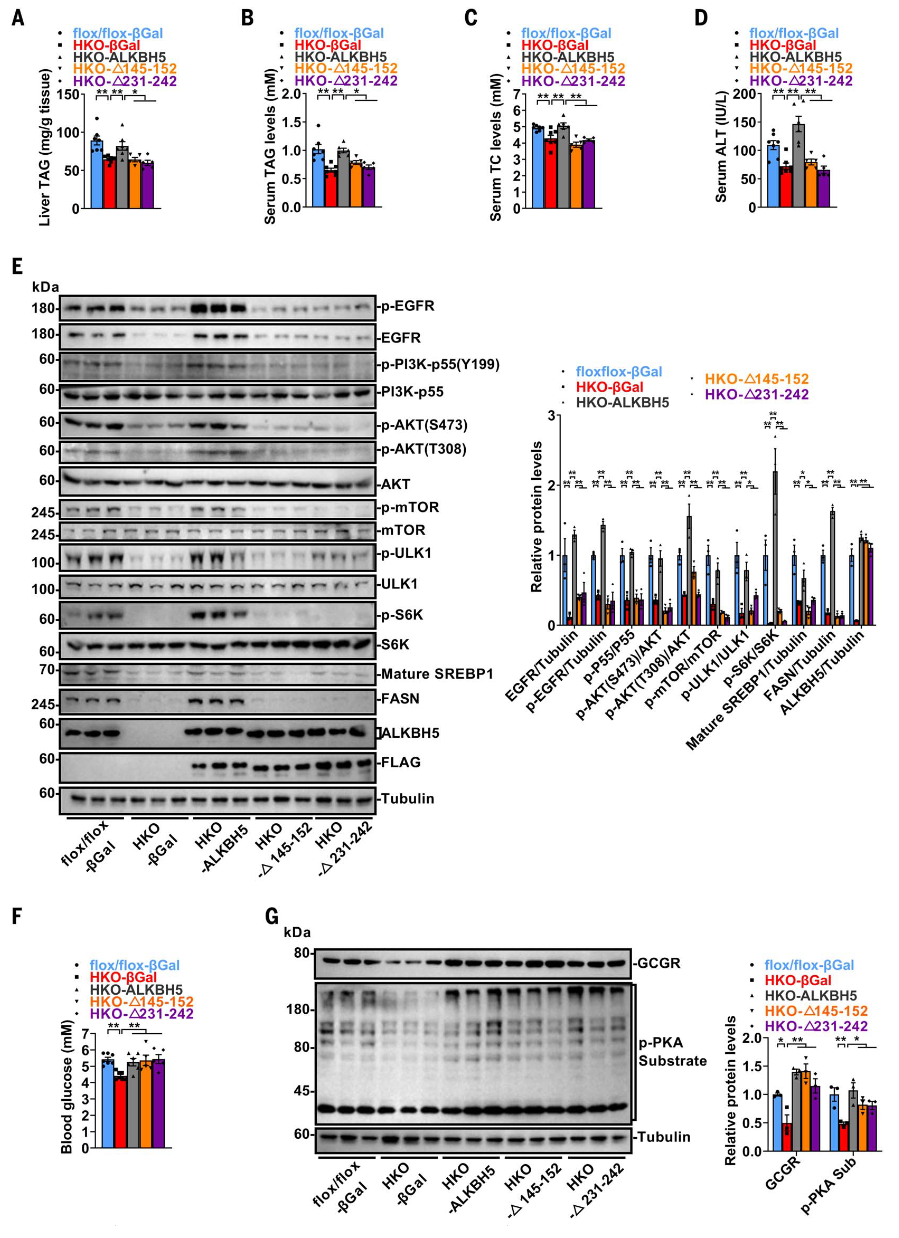
图7.Gln145-Gly152和Cys231-Glu242环对ALKBH5调控脂质稳态至关重要,但对葡萄糖稳态非必需
08
八、抑制肝脏ALKBH5的治疗潜力
肝细胞特异性缺失Alkbh5改善了葡萄糖和脂质稳态,表明ALKBH5是治疗代谢疾病的潜在靶点。通过RNA干扰技术,研究人员设计了AAV2/8-shRNA(AAV2/8-shAlkbh5-1和AAV2/8-shAlkbh5-2)以沉默db/db小鼠肝脏中的Alkbh5。两种AAV-shRNA均显著降低了肝脏ALKBH5水平。尽管db/db小鼠的体重未受影响,但肝脏Alkbh5的敲低导致空腹血糖降低、血清胰岛素水平下降、葡萄糖耐量改善、肝脏葡萄糖生成减少以及胰高血糖素敏感性降低,这些变化可能与GCGR-cAMP-PKA-CREB信号通路的下调及糖异生相关基因(G6pase和Pepck)表达减少有关。此外,Alkbh5敲低还改善了MAFLD,表现为肝脏TAG、血清TAG、TC和ALT活性降低,慢性炎症因子(如Tnfa、Ccl2、Il1a等)表达减少,这些改善可能与EGFR-PI3K-AKT-mTOR-ULK1-S6K-成熟SREBP1-CD36-FASN-SCD1信号通路的抑制有关。
为进一步验证肝脏ALKBH5抑制的治疗效果,研究人员使用GalNAc偶联的siRNA沉默db/db小鼠肝脏中的Alkbh5。GalNAc-siAlkbh5显著降低了db/db小鼠肝脏中的ALKBH5水平。尽管体重无显著差异,但GalNAc-siAlkbh5处理的小鼠表现出空腹血糖降低、血清胰岛素水平下降、葡萄糖耐量改善、肝脏葡萄糖生成减少以及胰高血糖素敏感性降低,这些变化与GCGR-cAMP-PKA-CREB信号通路的下调及糖异生相关基因表达减少相关。此外,GalNAc-siAlkbh5还改善了MAFLD,表现为肝脏TAG减少、脂滴减少、血清TAG和TC水平降低、ALT活性下降以及慢性炎症因子表达减少,这些改善可能与EGFR-PI3K-AKT-mTOR-ULK1-S6K-成熟SREBP1-CD36-FASN-SCD1信号通路的抑制有关。
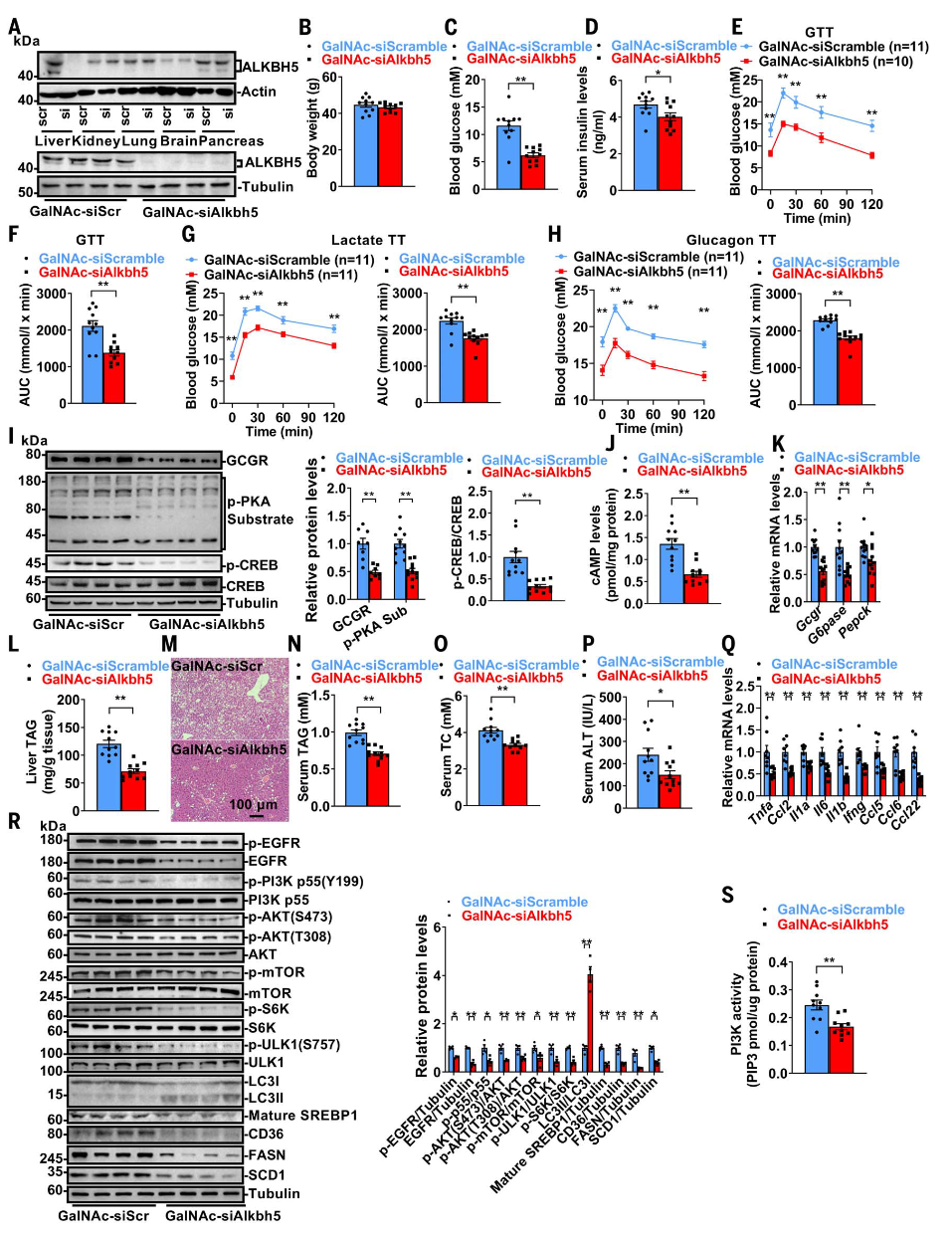
图8.肝脏特异性敲低Alkbh5逆转db/db小鼠的T2DM、MAFLD和高脂血症
ALKBH5在代谢疾病中通过两条独立信号通路调控糖脂稳态。一方面,ALKBH5通过Ser362磷酸化调控胰高血糖素-GCGR-cAMP-PKA-CREB信号通路,影响肝脏葡萄糖生成和糖异生相关基因表达,改善葡萄糖稳态。另一方面,ALKBH5通过结合Egfr内含子增强子,促进EGFR-PI3K-AKT-mTORC1信号通路,调控脂质合成相关基因,改善脂质稳态。Gln145-Gly152和Cys231-Glu242环对ALKBH5调控脂质稳态至关重要,但对葡萄糖稳态非必需。RNAi或GalNAc-siRNA介导的肝脏ALKBH5抑制可显著改善db/db小鼠的2型糖尿病和代谢MAFLD,表明ALKBH5是治疗代谢疾病的潜在靶点。
