

Cell:STING协调多发性硬化症中的神经元炎症应激反
时间 : 2024-10-25
多发性硬化症(MS)是中枢神经系统(CNS)最常见的自身免疫性疾病。在MS病程中,持续的炎症侵蚀神经元,神经变性和神经功能障碍加剧。免疫抑制剂虽能有效调节MS患者CNS浸润的免疫细胞,但不能干预进行性神经变性。因此,临床上仍需要直接作用于神经元的神经保护疗法来干预MS和其他神经退行性疾病中炎症诱导的神经变性。
对MS的广泛研究阐明了浸润的免疫细胞和CNS驻留的胶质细胞(特别是小胶质细胞和星形胶质细胞)在诱发慢性神经炎症中的重要作用。而神经元通常被认为是神经炎症刺激的被动接收者,主要由于其免疫相关基因表达水平低,对炎症刺激的形态变化和增殖能力有限。然而,最近逐渐认识到在神经炎症过程中,神经元在感知到多种细胞因子或应激源后启动神经元炎症应激反应(NISR),从而主动参与神经活动。
神经元不仅被动地响应细胞因子以维持稳态、协调发育,还能在炎症环境中主动调节免疫反应。干扰素-γ(IFNγ)是MS和其他神经退行性疾病CNS中高度富集的细胞因子,神经元对IFNγ的反应表明神经元积极参与CNS炎症。IFNγ与神经元上的Ⅱ型IFN受体结合,诱导STAT1表达和磷酸化,进而诱导干扰素应答基因(IRGs)。此外,IFNγ调节外周神经系统(PNS)中谷氨酸依赖性钙电流,表明细胞因子信号可改变神经元离子稳态。然而IFNγ的作用方式、对NISR的贡献、在炎症性神经退行性病变中的潜在作用仍未被探索。
除了细胞因子外,神经炎症期间的神经元稳态还受到神经元离子失衡的挑战。其主要原因是细胞外谷氨酸水平过高,其来源多样,包括死亡细胞释放谷氨酸,免疫细胞分泌谷氨酸活跃,神经胶质细胞(尤其是星形胶质细胞)谷氨酸代谢受损。过量的细胞外谷氨酸导致兴奋性毒性,这是由离子通道型谷氨酸受体激活后的细胞内钙积累介导的,也可能是由内部储存的钙释放介导的。这扰乱了神经元钙平衡,而钙平衡是神经元完整性、突触功能和存活的关键决定因素。
与神经元相反,免疫细胞等非兴奋性细胞主要依赖于钙库操纵性钙内流(SOCE)。这一过程起始于内质网(ER)上钙释放,促使内质网跨膜钙传感器蛋白——基质相互作用分子1和2(STIM1,STIM2)转运到内质网和质膜之间的接触位点。与钙释放-活化钙(CRAC)通道(如ORAI钙释放-活化钙调节剂1,ORAI1)聚集,使得钙从胞外流入并随后补充内质网储存钙。最近的研究发现,内质网钙释放也是神经炎症期间谷氨酸兴奋毒性细胞死亡的关键因素。然而,尽管内质网钙消耗(ERCa2+D)会加剧谷氨酸诱导的神经元钙积累,但其与炎症信号级联的相互作用以及CNS炎症期间STIM易位对神经元命运的影响仍不清楚。
2024年6月,来自德国汉堡-埃彭多夫大学医学中心的Manuel A. Friese团队在Cell上发表了题为“STING orchestrates the neuronal inflammatory stress response in multiple sclerosis”的研究文章。研究发现干扰素基因刺激因子(STING)作为炎性神经元损伤的关键介质,关联谷氨酸兴奋性毒性、神经元IFN信号传导和细胞死亡,而干预神经元STIM1-STING-GPX4信号可为多发性硬化症相关的神经退行性疾病提供新的治疗方向。
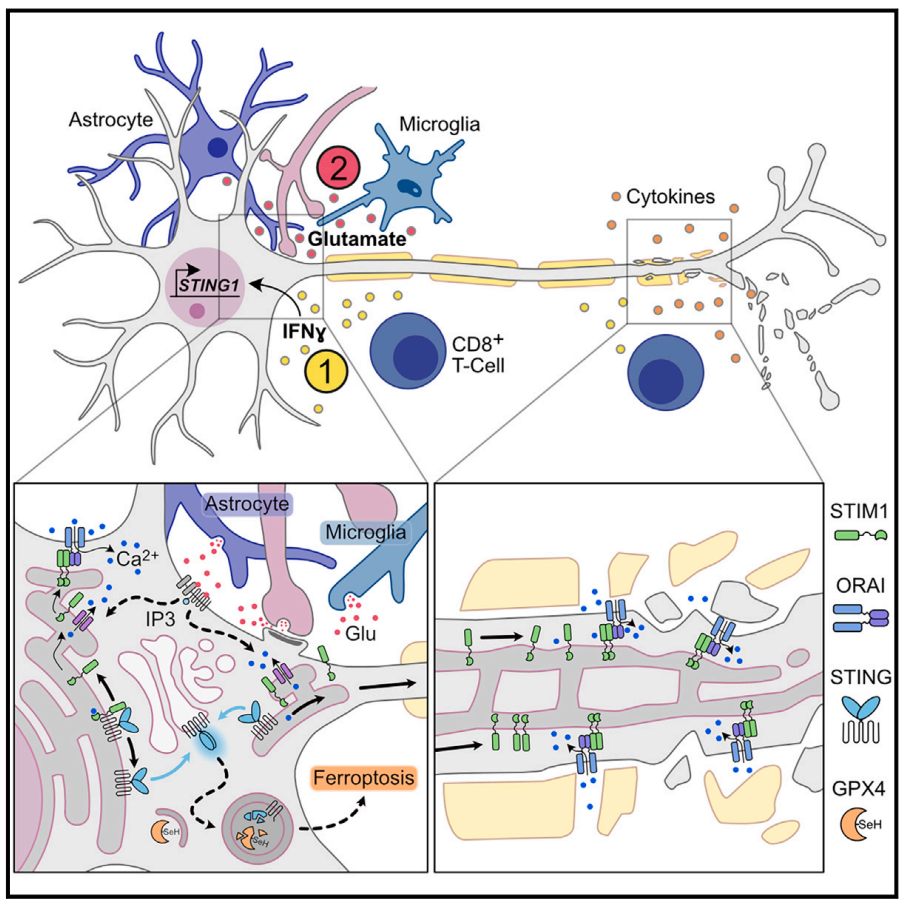 图解摘要
图解摘要

1.STIM1减轻炎症引起的神经变性
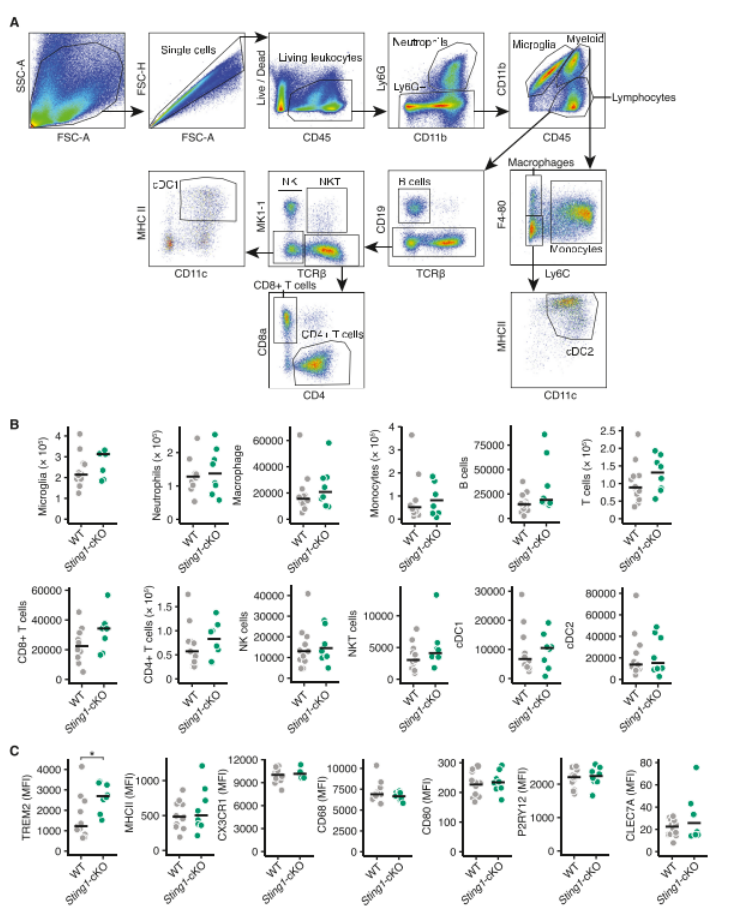
图S1 CNS炎症影响神经元STIM1的分布而不影响其表达
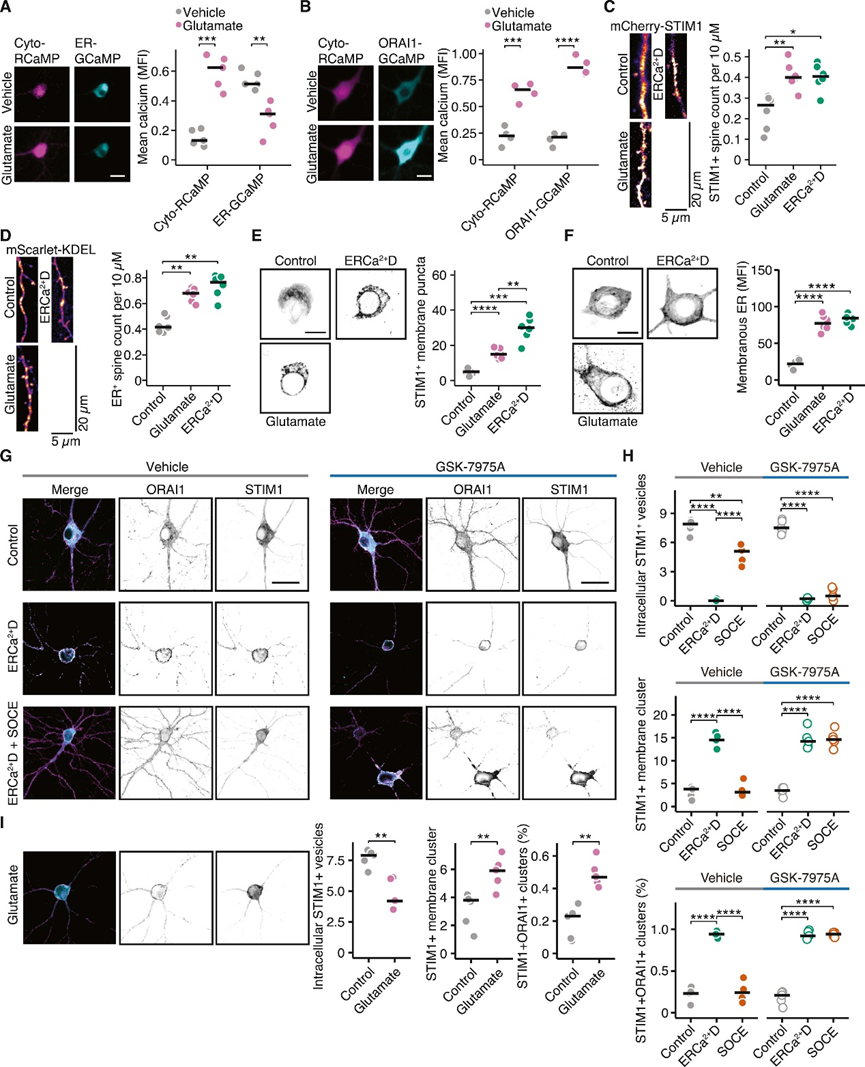
在证明STIM1减少或缺乏有助于EAE中炎症诱导的细胞死亡后,作者在体外评估了ERCa2+D在兴奋性毒性神经元细胞死亡中的作用。使用不同的基因编码钙传感器来监测不同神经元区室内谷氨酸诱导的钙变化。内质网钙耗竭的同时,谷氨酸兴奋毒性通过ORAI增加胞质钙水平和钙内流(图S2A-B)。同时,当神经元暴露于谷氨酸时,STIM1+ER+ 树突棘及STIM1+膜相关簇数量增加,靶向ERCa2+D亦是(图S2C-F)。值得注意的是,与ERCa2+D相比,谷氨酸兴奋毒性导致膜相关STIM1+簇与ORAI1共定位的显著增加。通过补充钙诱导SOCE,逆转STIM1+ORAI1+簇,可通过CRAC通道抑制剂GSK-7975A来阻断(图S2G-I)。因此,ERCa2+D是神经元对谷氨酸兴奋毒性应激反应的一部分,导致STIM1重新定位至细胞膜,与ORAI1相互作用。
为了从机制上解释Stim1缺失如何增加CNS炎症期间神经元的易感性,作者用谷氨酸刺激STIM1缺乏的皮层神经元,并测量细胞存活率和胞质钙积累。出乎意料的是,在缺乏Stim1的神经元和WT神经元之间,神经元易感性无差异,所以炎症环境可能是必需的(图S3A)。为了验证这一假设,将神经元长期暴露于IFNγ中。与WT神经元相比,这种炎症暴露使Stim1缺失神经元更容易发生兴奋毒性细胞死亡(图S3A)。谷氨酸诱导的胞质钙变化并不能解释其易感性差异(图S3B),这表明在Stim1-cKO EAE小鼠中观察到易感性增加需要炎性诱导因子。
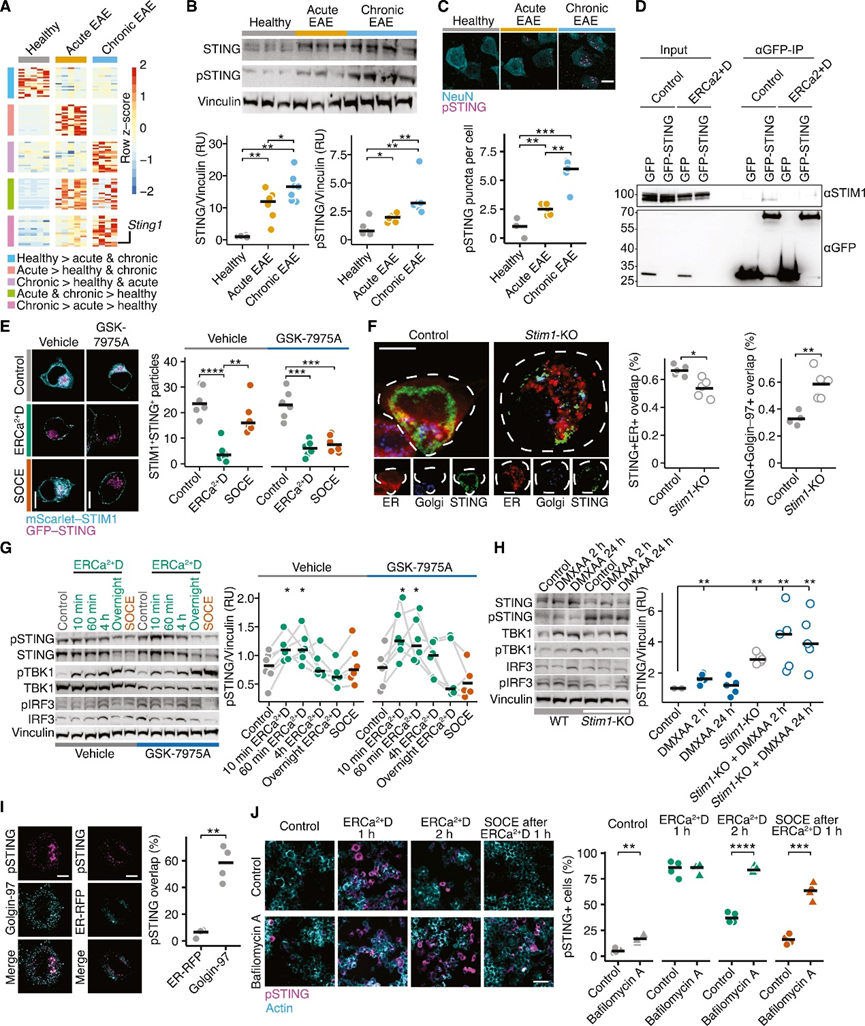
为了探索导致STIM1依赖性神经元易感性的炎症神经元反应,作者对急性和慢性EAE Glt25d2-EGFP/L10a小鼠皮层运动神经元的翻译核糖体亲和纯化(TRAP)谱进行了测序(图2A)。在急性和慢性EAE中最显著诱导的神经元基因之一是Sting1(图S3C),这是T细胞和成纤维细胞中STIM1的直接相互作用因子。STING是一种内质网跨膜蛋白,首次被报道为I型IFN对抗微生物感染的重要介质,在识别外源双链DNA(dsDNA)后被环GMP-AMP合成酶(cGAS)激活。然而,其在神经元中的表达和功能尚不清楚。此外,目前尚不清楚神经元中是否存在STING激活的替代途径。
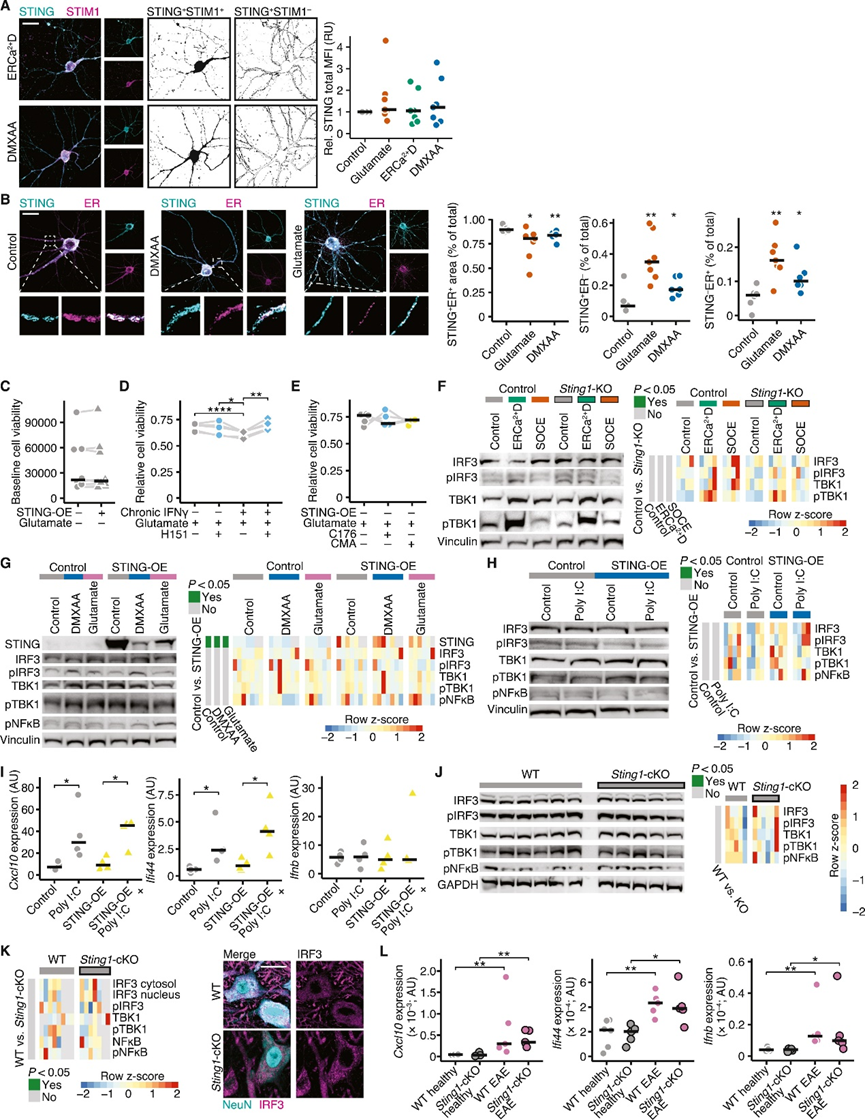
有趣的是,在正常细胞培养条件下,无法检测到原代神经元中STING的表达。相比之下,长时间暴露于IFNγ后,STING表达被显著诱导(图S3D),强调STING在炎症神经元中选择性表达。通过对总皮质裂解物的免疫印迹证实了其在体内的诱导作用,在急性和慢性EAE中,STING及其活化磷酸化形式(pSTING)持续增加(图2B)。这主要是由于神经元STING和pSTING水平的增加,在慢性疾病阶段达到峰值(图2C,S3E)。相反,急性EAE期间,非神经元细胞(特别是小胶质细胞)中STING和pSTING水平最高(图S3F-G)。
接着,作者探讨了经典STING信号的下游效应物在EAE中的表达。其中,只有干扰素调节因子3(IRF3)在EAE小鼠皮层中表达上调,可能由于IRF3在小胶质细胞中表达。尽管通过免疫印迹(图S3H)和免疫组化(图S3I)检测了IRF3、pIRF3、pTBK1和pNF-κB的神经元表达,但慢性EAE期间无增加。且pIRF3、pNF-kB或pTBK在非神经元细胞中不表达。以骨髓来源的树突状细胞(BMDCs)作为阳性对照,给予STING配DMXAA后显著激活典型的STING信号通路(图S3J)。因此,在CNS炎症期间,经典STING通路在皮层神经元中不被诱导。
因此,作者假设在CNS炎症期间,可能是神经元细胞体中STIM1的耗竭(可能易位到受损轴突)导致神经元中的STING激活。为了探究STING和STIM1是否存在相互作用,对STING过表达的Neuro-2a(N2a)细胞进行了免疫沉淀。内源性STIM1与EGFP标记的STING免疫共沉淀,表明STING和STIM1之间存在直接相互作用,ERCa2+D后这种相互作用显著降低(图2D)。在稳态下,EGFP-STING与mScarlet-STIM1在内质网共定位(图2E),在DMXAA化学激活STING后(图S3K)、ERCa2+D将STIM1易位至细胞膜(图2E)或基因敲除Stim1(图2F)后,STING转移到高尔基体。由于钙补充恢复了STIM-STING的共定位,因此SOCE可以调节STIM-STING的共定位,通过CRAC抑制剂GSK-7975A可抑制(图2E)。ERCa2+D使得STING与STIM分离,STING磷酸化(图2G)。此外,ERCa2+D或给予DMXAA或敲除Stim1增加了N2a细胞中的pSTING水平。值得注意的是,DMXAA并没有进一步增强Stim1缺失N2a细胞中的pSTING(图2H,S3L-M),且pSTING主要定位于高尔基体(图2I)。ERCa2+D诱导的STING磷酸化仅部分依赖于TBK1信号,因为TBK1抑制剂GSK-8612不能完全阻断pSTING(图S3N-O)。因此,神经元ERCa2+D导致STING转移至高尔基体,触发其磷酸化。
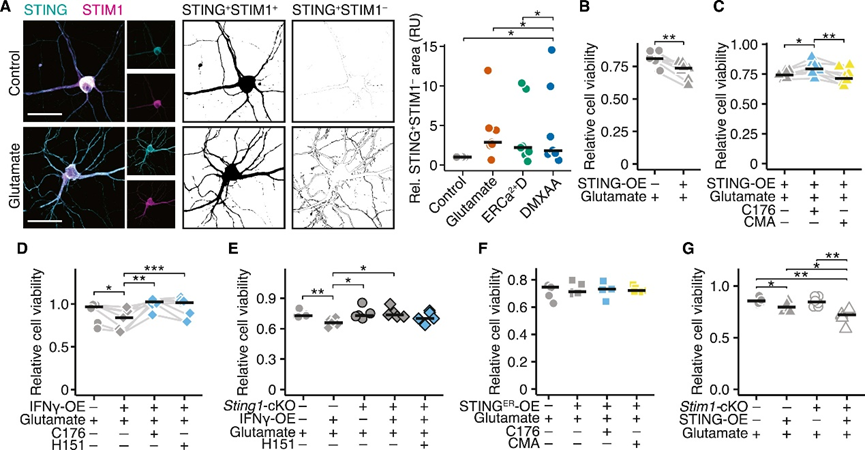
接着,作者探讨了STING如何影响体外神经元的活力。由于STING在稳态下不表达,可使用慢病毒表达STING,或长时间暴露于IFNγ或慢病毒表达Ifng来诱导STING(图S3D)。结果表明STING与STIM1在内质网共定位,并在靶向ERCa2+D、暴露于谷氨酸或DMXAA化学激活STING后共定位缺失(图3A,S4A-B)。表达STING的神经元和长期暴露于IFNγ的神经元更容易受到谷氨酸兴奋毒性的影响(图3B,S4D),可通过STING抑制剂C176或H151(图3C-D,S4D)或Sting1缺失神经元(图3E)来挽救,而用STING激动剂吖啶酮乙酸(CMA)并不能进一步增强毒性(图3C)。为了证实STING向高尔基体的易位是STING介导的易感性所必需的,对仅在ER保留表达STING(STINGER)的神经元开展相同实验(图S3K),发现抑制了神经元易感性的增加(图3F),未观察到C176的保护作用或CMA的任何影响,强调STING转运到高尔基体是增加神经元易损性所必需的。同样,在没有STING表达的神经元中,H151和C176都不能保护其免受兴奋性毒性的影响,突出其特异性(图S4D-E)。同样,与WT神经元相比,缺乏Stim1的神经元对STING诱导的细胞死亡更易感,证实了STIM1在炎症神经元ER中驻留STING的重要作用(图3G)。
为了了解STING介导的神经元易损的分子机制,作者首先分析了经典的STING信号通路。尽管ERCa2+D显著诱导STING和TBK1的磷酸化,但pTBK1的水平在WT和Sting1-KO的N2a细胞中相似(图S4F)。此外,与DMXAA处理的WT N2a细胞相比,Stim1-KO的N2a细胞显示出更高水平的pSTING,但没有激活STING的典型下游效应物(图2H,S3L)。与BMDCs等免疫细胞相比,神经元培养物在稳态下可检测到pTBK1、pIRF3和pNF-κB表达(图S3J)。通过免疫印迹检测,STING表达和暴露于DMXAA、poly(I:C)、谷氨酸或ERCa2+D,没有进一步增加这种表达(图S4G-H)。
同样,暴露于RNA模拟poly(I:C)可诱导神经元培养上清中Cxcl10和Ifi44的表达,而独立于STING表达(图S4I),强调神经元能够诱导独立于STING的IFN反应。为了验证STING是否在体内激活经典信号通路,作者首先分析了在WT和Sting1-cKO小鼠皮层中经典STING信号下游靶点的蛋白表达。再次观察到独立于STING的pTBK1和pIRF3高表达(图S4J)。通过免疫组化分析WT和Sting1-cKO小鼠EAE模型中的神经元也观察到这种现象(图S4K)。作者从WT或Sting-cKO健康或EAE小鼠中分离神经元核,比较Cxcl10、Ifi44和Ifnb的表达,与先前发现一致,EAE神经元中转录上调,独立于基因Sting1缺失(图S4L)。因此,作者证明了在体外或体内,经典的I型干扰素刺激途径在稳态下表达,并在CNS炎症过程中进一步上调,但不依赖于STING。
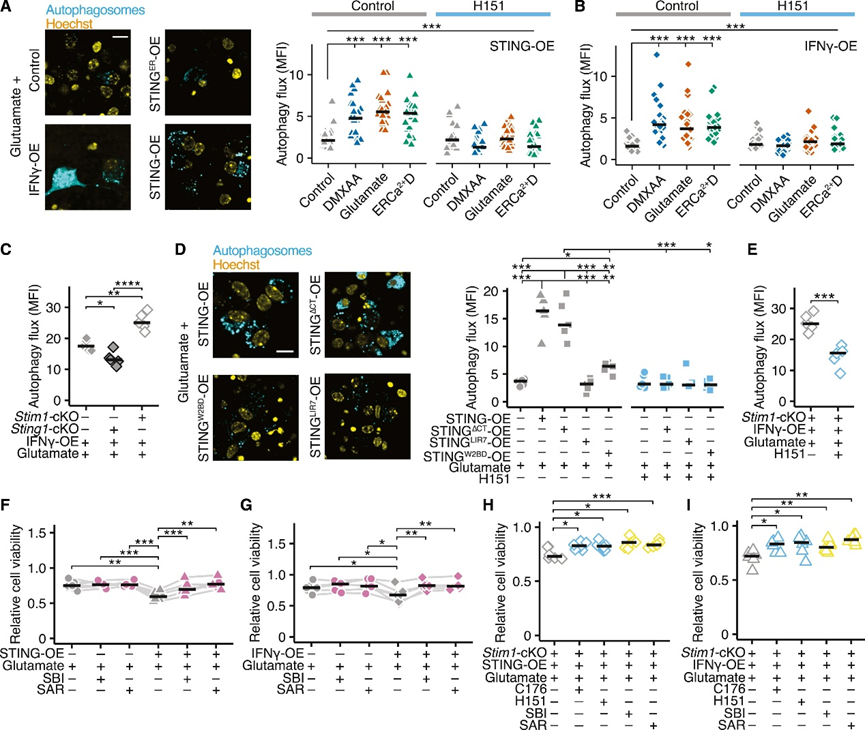
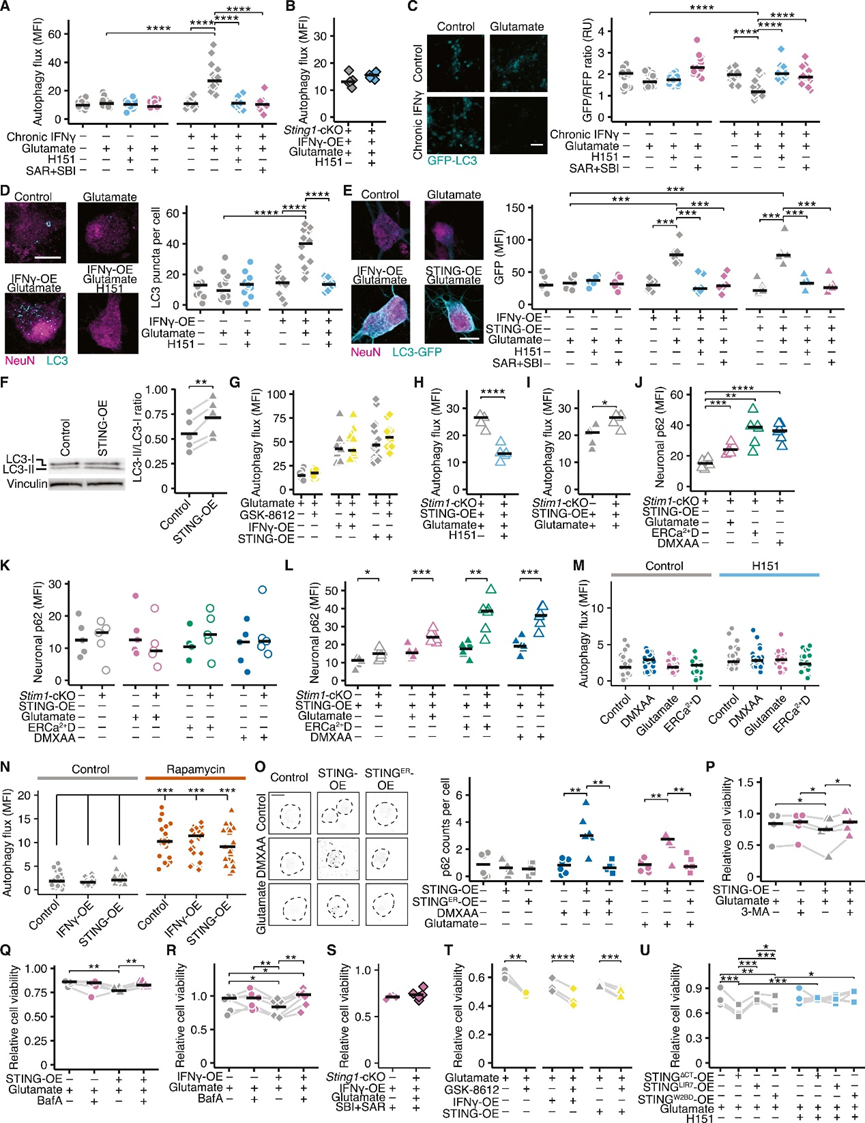
由于经典的STING信号通路在炎症神经元中不起作用,作者假设非经典的STING信号通路发挥作用,如诱导自噬。自噬是降解未折叠蛋白质、受损细胞器和病原体所必需的细胞过程。自噬诱导自噬体形成,将其内容物转运至溶酶体降解。虽然自噬是体内平衡和健康所必需的,但自噬改变逐渐被认为是衰老和多种疾病的诱因。STING介导的自噬通过促进病毒和细菌的清除来限制病原体感染。这一过程可独立于TBK1激活和IFN诱导而发生。用bafilomycin A1抑制溶酶体降解后对自噬泡进行染色,以评估表达STING(图4A)或暴露于IFNγ的神经元(图4B和S5A)的自噬通量。通过谷氨酸兴奋毒性,ERCa2+D诱导或DMXAA激活STING而诱导。事实上,导致神经元中STING激活的每一种刺激都能诱导自噬,可通过H151或基因敲除Sting1来抑制(图4C,S5B)。通过多种方式进一步证实了谷氨酸处理后表达STING或暴露于IFNγ的神经元自噬增加(图S5C-D)。此外,在表达STING的神经元中,LC3-II/LC3-I比值升高(图S5F)。但与经典STING信号通路无关,因为使用TBK1抑制剂自噬通量无显著变化(图S5G),而C端截短的STINGΔCT突变体(不能与TBK1和IRF3相互作用)自噬通量增加(图4D)。相比之下,LC3相互作用7基序(STINGLIR7)或WD重复结构域磷酸肌醇相互作用蛋白2(WIPI2;STINGW2BD)(两者是STING-LC3相互作用所需)的缺失突变体中检测到较少的自噬通量(图4D)。但在表达STINGW2BD的神经元中,谷氨酸诱导的自噬通量明显高于表达STINGLIR7的神经元,表明除了WIPI2之外,STING诱导的神经元自噬还需要其他因素。
此外,Stim1缺失神经元的STING依赖性自噬进一步增加(图4C,4E,S5H-L)。给予雷帕霉素后自噬通量和最大自噬能力的基线水平在所有条件下均相似(图S5M-N)。为了评估STING依赖性自噬中从内质网释放STING的必要性,将表达STING的神经元与表达STINGER的神经元相比较。发现表达STING而非STINGER的神经元中,p62+ puncta(自噬小体)增加(图S5O)。以上数据表明,神经元中非经典自噬STING通路被激活。
为了检验增加的自噬通量是否诱导STING介导的神经元对谷氨酸的易感,使用了各种自噬抑制剂处理表达STING的神经元。事实上,抑制剂3-methyladenine、bafilomycin A1、SAR405(SAR)和SBI0206965(SBI)在WT(图4F-G,S5P-R)和Stim1缺失神经元(图4H-I)中逆转了STING诱导的神经元易感性。在没有STING表达的情况下,这些抑制剂不影响神经元的活力,也不为缺乏Sting1的神经元提供额外的保护(图5S)。此外,STINGΔCT的表达增加了神经元对谷氨酸的易感性。当神经元表达STINGW2BD时,易感性显著降低,而表达STINGLIR7的神经元不存在该现象,药理学上抑制TBK1时易感性加剧(图S5T-U)。这共同表明,在炎症条件下,ERCa2+D激活的STING通过诱导自噬来放大神经元损伤。
图S5 抑制自噬可防止STING诱导的神经元损伤
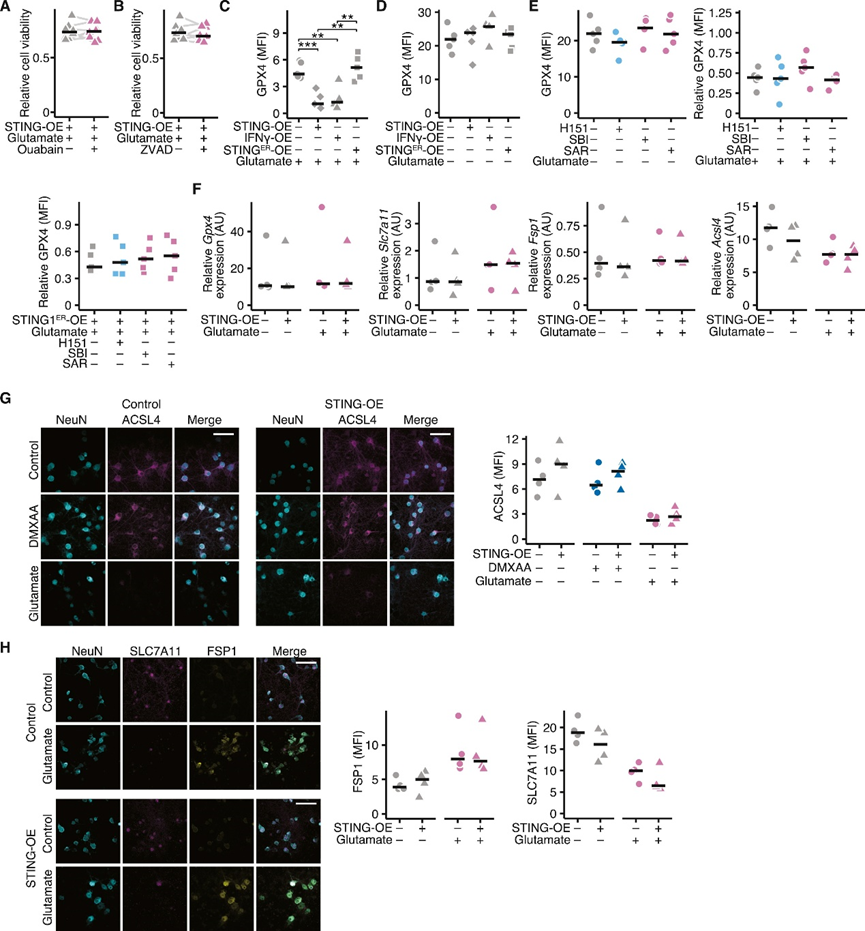
自噬依赖性细胞死亡的两种不同机制已经在癌细胞中被描述,而在神经元中尚未被探索。自噬可以激活Na+/K+-ATP酶依赖性细胞凋亡,即自体死亡autosis。然而,使用ouabain阻断Na+/K+-ATP酶(图S6A)或使用pan-caspase抑制剂Z-VAD-FMK(图S6B)并不能逆转表达STING的神经元对谷氨酸易感性增加。这表明autosis不是STING诱导的神经元自噬依赖性细胞死亡的原因。
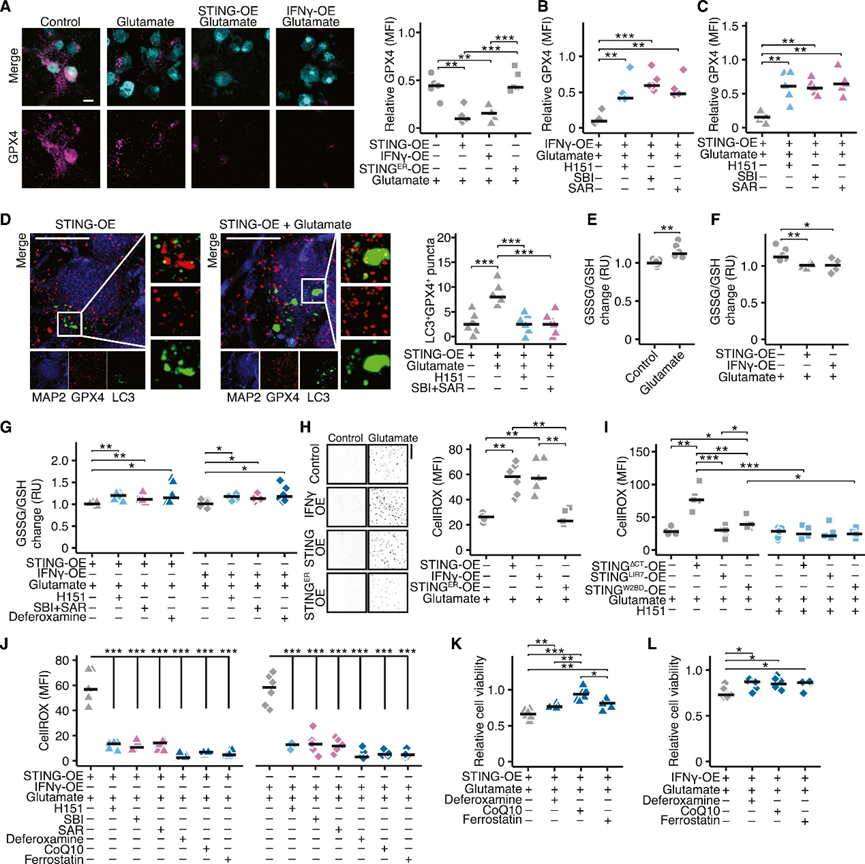
因此作者专注于第二种机制:铁死亡,一种铁依赖的细胞死亡途径。这一途径通过谷胱甘肽过氧化物酶4(GPX4)的自噬降解在癌细胞中被显著诱导。GPX4可防止脂质过氧化并与铁死亡有关。暴露于谷氨酸后,神经元细胞体和树突中的GPX4水平迅速下降,表达STING的神经元中GPX4下降水平明显高于STINGER(图5A,S6C)。可用STING抑制剂H151和自噬抑制剂SAR和SBI部分挽救(图5B-C)。在谷氨酸兴奋毒性诱导STING表达的神经元中,GPX4+LC3+斑点表明GPX4阳性的自噬体数量增加,说明自噬驱动GPX4降解。该作用被药理学抑制STING或自噬所逆转(图5D)。在过表达STING的神经元中,GPX4的基线水平没有差异;在对照或表达STINGER的神经元中,H151、SAR或SBI也没有挽救谷氨酸诱导的GPX4减少(图S6D-E)。此外,其他已知的铁死亡关键调节因子,长链酰基辅酶A合成酶4(ACSL4),溶质载体家族成员(SLC7A11),铁死亡抑制蛋白1(FSP1),在稳态或谷氨酸刺激后表达没有差异(图S6F-H)。这些结果表明,在炎症和兴奋性毒性作用下,STING可以诱导神经元中GPX4的自噬降解。
接下来,作者探讨了GPX4自噬降解在神经元中的功能相关性。首先,测量了氧化型谷胱甘肽(GSSG)和总谷胱甘肽(GSH)的比值。GPX4活性决定性地调节GSSG/GSH比值,因为GPX4通过将两分子还原型GSH氧化为GSSG而将磷脂氢过氧化物还原为相应的脂醇。谷氨酸暴露增加了GSSG/GSH比率(图5E),降低了总GSH水平(图S7A),证明了兴奋毒性谷氨酸应激下神经元中GPX4的酶活性。但在表达STING的神经元中,谷氨酸刺激后GSSG/GSH比值不再增加(图5F),表明GPX4活性降低。给予神经元H151、SBI或SAR以及抗氧化剂去铁胺后,GSSG/GSH比值恢复(图5G)。在表达STING的神经元中,基线GSSG/GSH相当,并且谷氨酸诱导的总GSH降低不受神经元STING表达的影响(图S7B-E)。与对照或表达STINGER的神经元相比,在谷氨酸暴露后,表达STING的神经元表现出更高的ROS水平(图5H,S7F)。STINGΔCT显著增加ROS产生,而表达STINGW2BD的神经元中ROS少得多(图5I),进一步证实了独立于经典的STING途径。H151、SBI和SAR以及铁螯合剂去铁胺和辅酶Q10(CoQ10)和铁抑素处理可以挽救ROS的产生,但TBK1抑制剂GSK-8612则不能(图5J,S7G-J)。因此,STING通过GPX4的自噬降解干扰炎症神经元的适应性抗氧化反应。由于Gpx4缺失导致铁死亡激活,作者假设STING在炎症诱导的神经元铁死亡中发挥关键调节作用。在WT(图5K-L,S7K-L)和Stim1缺失神经元(图S7M-O)中,用抗氧化剂去铁胺、CoQ10和铁抑素处理可保护表达STING的神经元免受谷氨酸兴奋毒性的影响。因此,STING是神经炎症中神经元自噬依赖性铁死亡的关键调节因子。
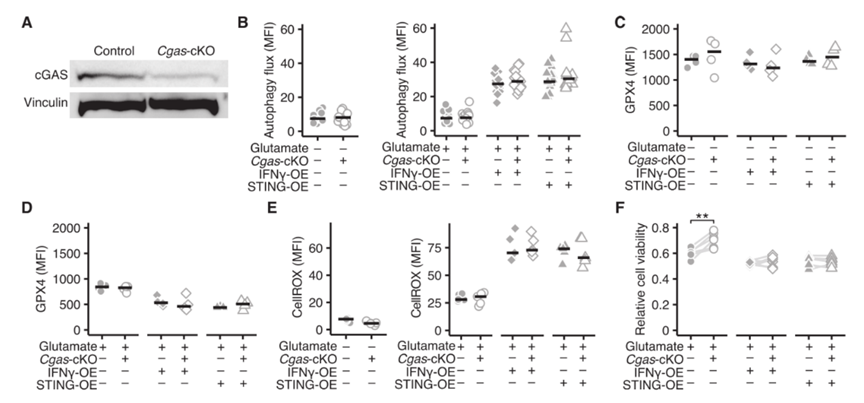
由于无法确定是否需要稳态cGAS活性来产生低水平cGAMP,从而持续驱动STING运输和信号传导,作者探索了cGAS是否也参与谷氨酸诱导的STING介导的神经元铁死亡。作者构建了缺失Cgas的神经元(图S8A),并通过慢病毒转染或慢性暴露于IFNγ诱导STING表达。在表达STING的神经元中,谷氨酸兴奋毒性诱导的自噬通量增加(图S8B)、GPX4降解(图S8C-D)和ROS产生(图S8E)在WT和缺失Cgas的神经元中是相似的。不表达STING的cGAS缺陷神经元对谷氨酸兴奋毒性的敏感性较低,表明在没有STING的情况下,cGAS在神经元中具有明显的生理作用(图S8F)。因此,由谷氨酸兴奋性毒性引起的STING诱导的铁死亡可能不依赖于神经元中的cGAS。

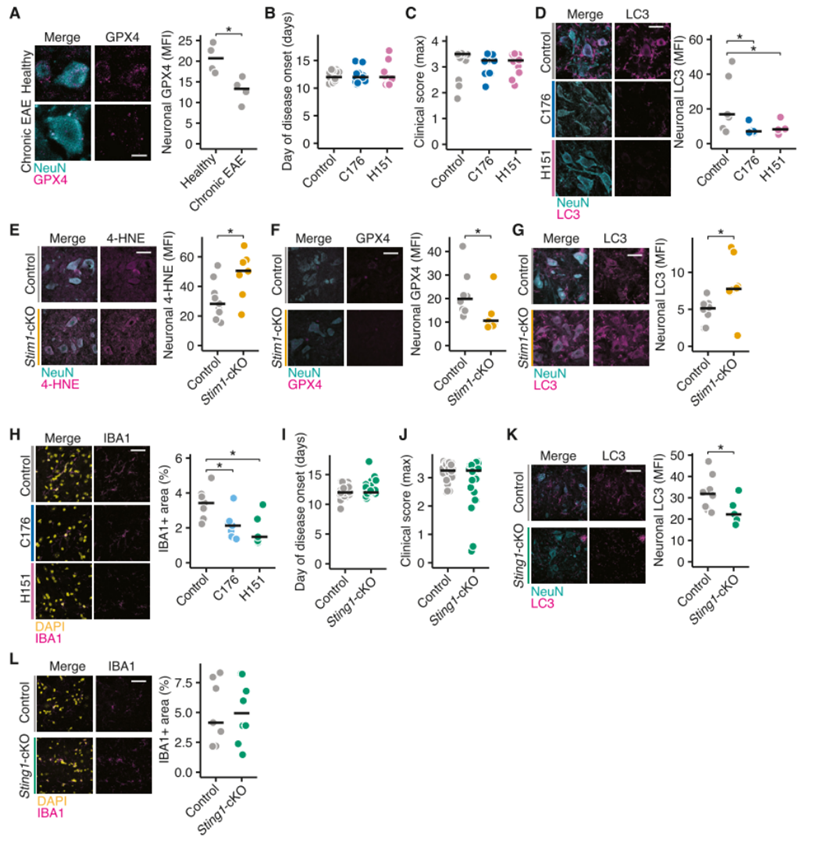
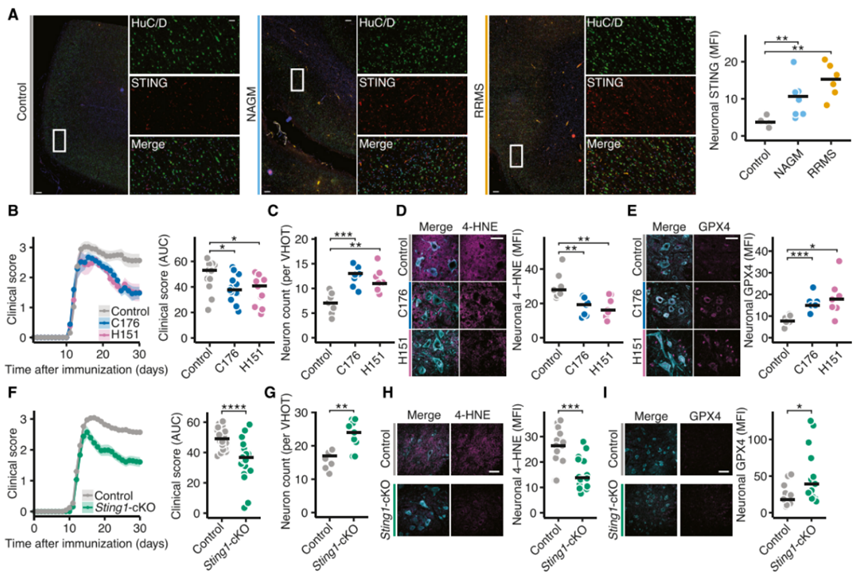
由于GPX4在pwMS和EAE神经元中下调(图S9A),作者探索了抑制STING作为治疗MS神经变性的治疗策略。首先,作者分析了pwMS大脑神经元STING的表达。与EAE类似,在进展性MS患者的正常灰质(NAGM)皮层神经元以及复发缓解型MS患者和活动性炎症患者的皮层神经元STING表达上调(图6A)。鉴于STING在神经元的广泛上调,将其定位为治疗干预的预期靶点,使用STING拮抗剂C176或H151治疗EAE动物。从发病开始每日治疗可降低临床疾病评分(图6B,S9B-C),减少神经元丢失(图6C)。用STING拮抗剂治疗的小鼠显示出较低水平的自噬标志物LC3(图S9D)和铁死亡标志物4-HNE(图6D),神经元中GPX4表达增加(图6E)。相比之下,Stim1-cKO EAE小鼠显示较低的GPX4水平和较高的LC3和4-HNE水平,支持自噬依赖性铁死亡在缺乏STIM1时加剧(图S9E-G)。因此,药理STING拮抗剂可减少炎症诱导的神经元铁死亡。
然而,正如STING能诱导小胶质细胞强激活那样,作者发现经治疗的EAE小鼠中激活的小胶质细数量减少(图S9H)。为了绕过STING拮抗剂对小胶质细胞活化的抑制作用,作者在神经元特异性的Sting-cKO小鼠上构建了EAE。结果表明Sting-cKO小鼠病程改善(图6F,S9I-J),神经元数量增加(图6G),免疫荧光结果表明神经元LC3(图S9K)和4-HNE(图6H)表达下调,GPX4水平上调(图6I)。且没有观察到WT和Sting-cKO EAE小鼠小胶质细胞和CNS浸润免疫细胞的数量存在差异(图S9L,S10A-B)。总的来说,作者的数据表明抑制STING是在神经炎症条件下直接干预NISR的杰出治疗范例,发挥神经保护作用。
对MS的广泛研究阐明了浸润的免疫细胞和CNS驻留的胶质细胞(特别是小胶质细胞和星形胶质细胞)在诱发慢性神经炎症中的重要作用。而神经元通常被认为是神经炎症刺激的被动接收者,主要由于其免疫相关基因表达水平低,对炎症刺激的形态变化和增殖能力有限。然而,最近逐渐认识到在神经炎症过程中,神经元在感知到多种细胞因子或应激源后启动神经元炎症应激反应(NISR),从而主动参与神经活动。
神经元不仅被动地响应细胞因子以维持稳态、协调发育,还能在炎症环境中主动调节免疫反应。干扰素-γ(IFNγ)是MS和其他神经退行性疾病CNS中高度富集的细胞因子,神经元对IFNγ的反应表明神经元积极参与CNS炎症。IFNγ与神经元上的Ⅱ型IFN受体结合,诱导STAT1表达和磷酸化,进而诱导干扰素应答基因(IRGs)。此外,IFNγ调节外周神经系统(PNS)中谷氨酸依赖性钙电流,表明细胞因子信号可改变神经元离子稳态。然而IFNγ的作用方式、对NISR的贡献、在炎症性神经退行性病变中的潜在作用仍未被探索。
除了细胞因子外,神经炎症期间的神经元稳态还受到神经元离子失衡的挑战。其主要原因是细胞外谷氨酸水平过高,其来源多样,包括死亡细胞释放谷氨酸,免疫细胞分泌谷氨酸活跃,神经胶质细胞(尤其是星形胶质细胞)谷氨酸代谢受损。过量的细胞外谷氨酸导致兴奋性毒性,这是由离子通道型谷氨酸受体激活后的细胞内钙积累介导的,也可能是由内部储存的钙释放介导的。这扰乱了神经元钙平衡,而钙平衡是神经元完整性、突触功能和存活的关键决定因素。
与神经元相反,免疫细胞等非兴奋性细胞主要依赖于钙库操纵性钙内流(SOCE)。这一过程起始于内质网(ER)上钙释放,促使内质网跨膜钙传感器蛋白——基质相互作用分子1和2(STIM1,STIM2)转运到内质网和质膜之间的接触位点。与钙释放-活化钙(CRAC)通道(如ORAI钙释放-活化钙调节剂1,ORAI1)聚集,使得钙从胞外流入并随后补充内质网储存钙。最近的研究发现,内质网钙释放也是神经炎症期间谷氨酸兴奋毒性细胞死亡的关键因素。然而,尽管内质网钙消耗(ERCa2+D)会加剧谷氨酸诱导的神经元钙积累,但其与炎症信号级联的相互作用以及CNS炎症期间STIM易位对神经元命运的影响仍不清楚。
2024年6月,来自德国汉堡-埃彭多夫大学医学中心的Manuel A. Friese团队在Cell上发表了题为“STING orchestrates the neuronal inflammatory stress response in multiple sclerosis”的研究文章。研究发现干扰素基因刺激因子(STING)作为炎性神经元损伤的关键介质,关联谷氨酸兴奋性毒性、神经元IFN信号传导和细胞死亡,而干预神经元STIM1-STING-GPX4信号可为多发性硬化症相关的神经退行性疾病提供新的治疗方向。
图解摘要
为了研究神经元STIM蛋白是否参与CNS炎症期间的神经变性,作者构建了神经元特异性缺失Stim1或Stim2的小鼠(Stim1-cKO/Stim2-cKO)。用MOG35-55肽主动免疫小鼠构建实验性自身免疫性脑脊髓炎(EAE)模型。Stim1-cKO(图1A-B,S1A-B)而非Stim2-cKO(图1C-D,S1C-D)显示出病程加重,神经元损失增加,突出表明STIM1是神经元抵抗神经炎症所必需的。为了了解STIM1在神经炎症中的具体作用,作者首先表征了STIM1在EAE中的表达和亚细胞定位。在EAE小鼠皮层和接受脑活检的MS患者(pwMS)的神经元胞体中检测到STIM1蛋白的显著减少(图1E-F)。这种减少不是由于转录调节,因为神经元Stim1mRNA在EAE中没有表达差异(图1G)。由于神经元的ERCa2+D在EAE动物和兴奋毒性细胞死亡中显著,作者假设EAE神经元中SOCE的激活可能是突触和轴突中STIM1水平代偿性增加导致的。为了探讨这一点,作者首先比较了急性和慢性EAE小鼠皮质突触神经小体中的STIM1蛋白水平(图S1E)。有趣的是,慢性EAE模型突触神经小体的STIM1蛋白水平升高,而整个皮层的STIM1蛋白水平则降低(图1H)。接着,作者分析了这些皮层运动神经元到脊髓脊柱的轴突投射。轴突STIM1染色在急性和慢性疾病阶段升高(图1I,S1F),支持了作者假设,即SOCE激活后突触和轴突中STIM1增加。鉴于轴突中STIM1的增加可能反映了兴奋性毒性损伤,作者接下来测试了STIM1是否优先定位于受损轴突部位。为此,作者比较了健康轴突磷酸化(SMI31+)和受损轴突非磷酸化(SMI32+)神经丝重链蛋白中的STIM1水平。事实上,与未损伤或恢复的SMI31+轴突相比,在慢性EAE期间,损伤的SMI32+轴突中STIM1水平明显更高(图1J,S1G)。值得注意的是,脊髓前角运动神经元中Stim1mRNA和STIM1蛋白水平没有差异(图S1H-I)。总之,Stim1缺失增加了神经元对炎症性神经变性的易感性。此外,CNS炎症与神经元细胞体中STIM1的显著减少有关,这可能是由于STIM1易位到受损轴突或这些轴突中STIM1代偿性增加。
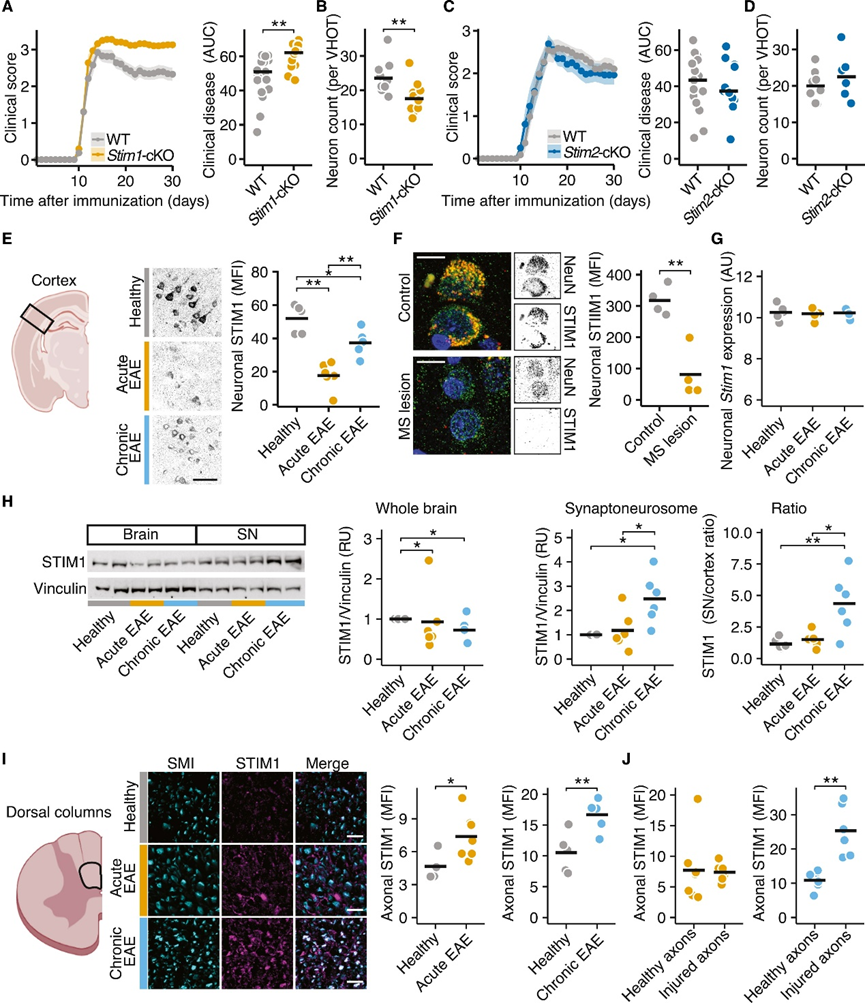
图1 STIM1缺失增加CNS炎症中的神经元易感性
图1 STIM1缺失增加CNS炎症中的神经元易感性
2.谷氨酸诱导的兴奋性毒性激活SOCE
图S2 谷氨酸兴奋毒性激活神经元中的SOCE
3.神经元STING由SOCE激活
为了探索导致STIM1依赖性神经元易感性的炎症神经元反应,作者对急性和慢性EAE Glt25d2-EGFP/L10a小鼠皮层运动神经元的翻译核糖体亲和纯化(TRAP)谱进行了测序(图2A)。在急性和慢性EAE中最显著诱导的神经元基因之一是Sting1(图S3C),这是T细胞和成纤维细胞中STIM1的直接相互作用因子。STING是一种内质网跨膜蛋白,首次被报道为I型IFN对抗微生物感染的重要介质,在识别外源双链DNA(dsDNA)后被环GMP-AMP合成酶(cGAS)激活。然而,其在神经元中的表达和功能尚不清楚。此外,目前尚不清楚神经元中是否存在STING激活的替代途径。
有趣的是,在正常细胞培养条件下,无法检测到原代神经元中STING的表达。相比之下,长时间暴露于IFNγ后,STING表达被显著诱导(图S3D),强调STING在炎症神经元中选择性表达。通过对总皮质裂解物的免疫印迹证实了其在体内的诱导作用,在急性和慢性EAE中,STING及其活化磷酸化形式(pSTING)持续增加(图2B)。这主要是由于神经元STING和pSTING水平的增加,在慢性疾病阶段达到峰值(图2C,S3E)。相反,急性EAE期间,非神经元细胞(特别是小胶质细胞)中STING和pSTING水平最高(图S3F-G)。
接着,作者探讨了经典STING信号的下游效应物在EAE中的表达。其中,只有干扰素调节因子3(IRF3)在EAE小鼠皮层中表达上调,可能由于IRF3在小胶质细胞中表达。尽管通过免疫印迹(图S3H)和免疫组化(图S3I)检测了IRF3、pIRF3、pTBK1和pNF-κB的神经元表达,但慢性EAE期间无增加。且pIRF3、pNF-kB或pTBK在非神经元细胞中不表达。以骨髓来源的树突状细胞(BMDCs)作为阳性对照,给予STING配DMXAA后显著激活典型的STING信号通路(图S3J)。因此,在CNS炎症期间,经典STING通路在皮层神经元中不被诱导。
因此,作者假设在CNS炎症期间,可能是神经元细胞体中STIM1的耗竭(可能易位到受损轴突)导致神经元中的STING激活。为了探究STING和STIM1是否存在相互作用,对STING过表达的Neuro-2a(N2a)细胞进行了免疫沉淀。内源性STIM1与EGFP标记的STING免疫共沉淀,表明STING和STIM1之间存在直接相互作用,ERCa2+D后这种相互作用显著降低(图2D)。在稳态下,EGFP-STING与mScarlet-STIM1在内质网共定位(图2E),在DMXAA化学激活STING后(图S3K)、ERCa2+D将STIM1易位至细胞膜(图2E)或基因敲除Stim1(图2F)后,STING转移到高尔基体。由于钙补充恢复了STIM-STING的共定位,因此SOCE可以调节STIM-STING的共定位,通过CRAC抑制剂GSK-7975A可抑制(图2E)。ERCa2+D使得STING与STIM分离,STING磷酸化(图2G)。此外,ERCa2+D或给予DMXAA或敲除Stim1增加了N2a细胞中的pSTING水平。值得注意的是,DMXAA并没有进一步增强Stim1缺失N2a细胞中的pSTING(图2H,S3L-M),且pSTING主要定位于高尔基体(图2I)。ERCa2+D诱导的STING磷酸化仅部分依赖于TBK1信号,因为TBK1抑制剂GSK-8612不能完全阻断pSTING(图S3N-O)。因此,神经元ERCa2+D导致STING转移至高尔基体,触发其磷酸化。
由于观察到ERCa2+D只导致pSTING的短暂增加,并且其随后的降解既不会被SOCE激活进一步增强,也不会被GSK-7975A阻断(图2G),作者假设ERCa2+D特异性地增加了STING信号的启动而不影响其降解。由于最近有证据表明STING在溶酶体中持续降解,用溶酶体功能抑制剂bafilomycin A处理N2a细胞,并测量ERCa2+D和随后SOCE激活后的pSTING水平。无论是否存在SOCE,溶酶体抑制并不增加ERCa2+D诱导的STING磷酸化,但阻止了pSTING下调(图2J)。因此,ERCa2+D增强神经元中STING的激活,并通过溶酶体降解终止,与钙补充无关。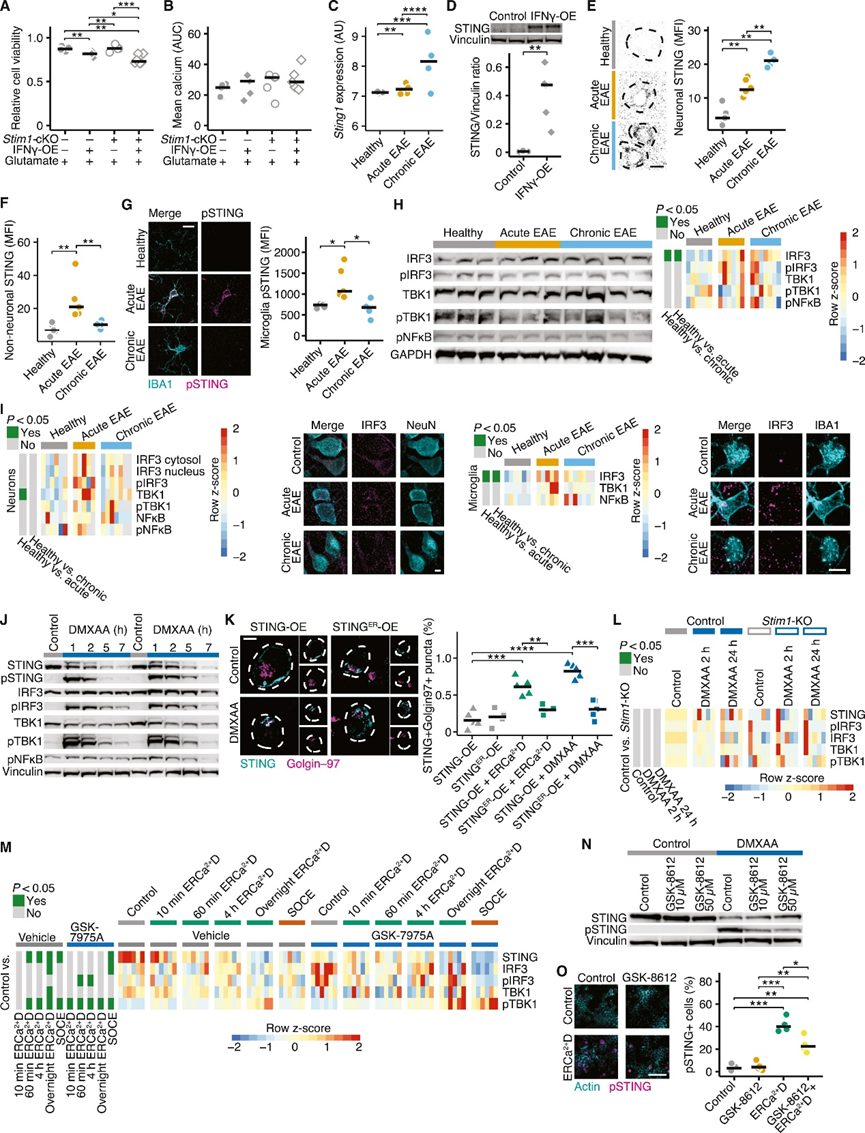
图S3 SOCE激活STING
图2 内质网钙耗竭诱导STING转移至高尔基体并促进其激活
4.神经元中STING不诱导经典通路
为了了解STING介导的神经元易损的分子机制,作者首先分析了经典的STING信号通路。尽管ERCa2+D显著诱导STING和TBK1的磷酸化,但pTBK1的水平在WT和Sting1-KO的N2a细胞中相似(图S4F)。此外,与DMXAA处理的WT N2a细胞相比,Stim1-KO的N2a细胞显示出更高水平的pSTING,但没有激活STING的典型下游效应物(图2H,S3L)。与BMDCs等免疫细胞相比,神经元培养物在稳态下可检测到pTBK1、pIRF3和pNF-κB表达(图S3J)。通过免疫印迹检测,STING表达和暴露于DMXAA、poly(I:C)、谷氨酸或ERCa2+D,没有进一步增加这种表达(图S4G-H)。
同样,暴露于RNA模拟poly(I:C)可诱导神经元培养上清中Cxcl10和Ifi44的表达,而独立于STING表达(图S4I),强调神经元能够诱导独立于STING的IFN反应。为了验证STING是否在体内激活经典信号通路,作者首先分析了在WT和Sting1-cKO小鼠皮层中经典STING信号下游靶点的蛋白表达。再次观察到独立于STING的pTBK1和pIRF3高表达(图S4J)。通过免疫组化分析WT和Sting1-cKO小鼠EAE模型中的神经元也观察到这种现象(图S4K)。作者从WT或Sting-cKO健康或EAE小鼠中分离神经元核,比较Cxcl10、Ifi44和Ifnb的表达,与先前发现一致,EAE神经元中转录上调,独立于基因Sting1缺失(图S4L)。因此,作者证明了在体外或体内,经典的I型干扰素刺激途径在稳态下表达,并在CNS炎症过程中进一步上调,但不依赖于STING。
图3 STING增加神经元对兴奋性毒性的易感性
图S4 经典的STING通路在神经元中未被激活
5.STING诱导神经元自噬依赖性细胞死亡
此外,Stim1缺失神经元的STING依赖性自噬进一步增加(图4C,4E,S5H-L)。给予雷帕霉素后自噬通量和最大自噬能力的基线水平在所有条件下均相似(图S5M-N)。为了评估STING依赖性自噬中从内质网释放STING的必要性,将表达STING的神经元与表达STINGER的神经元相比较。发现表达STING而非STINGER的神经元中,p62+ puncta(自噬小体)增加(图S5O)。以上数据表明,神经元中非经典自噬STING通路被激活。
图4 STING激活神经元中自噬依赖的细胞死亡
6.STING诱导的自噬通过降解GPX4激活铁死亡
接下来,作者探讨了GPX4自噬降解在神经元中的功能相关性。首先,测量了氧化型谷胱甘肽(GSSG)和总谷胱甘肽(GSH)的比值。GPX4活性决定性地调节GSSG/GSH比值,因为GPX4通过将两分子还原型GSH氧化为GSSG而将磷脂氢过氧化物还原为相应的脂醇。谷氨酸暴露增加了GSSG/GSH比率(图5E),降低了总GSH水平(图S7A),证明了兴奋毒性谷氨酸应激下神经元中GPX4的酶活性。但在表达STING的神经元中,谷氨酸刺激后GSSG/GSH比值不再增加(图5F),表明GPX4活性降低。给予神经元H151、SBI或SAR以及抗氧化剂去铁胺后,GSSG/GSH比值恢复(图5G)。在表达STING的神经元中,基线GSSG/GSH相当,并且谷氨酸诱导的总GSH降低不受神经元STING表达的影响(图S7B-E)。与对照或表达STINGER的神经元相比,在谷氨酸暴露后,表达STING的神经元表现出更高的ROS水平(图5H,S7F)。STINGΔCT显著增加ROS产生,而表达STINGW2BD的神经元中ROS少得多(图5I),进一步证实了独立于经典的STING途径。H151、SBI和SAR以及铁螯合剂去铁胺和辅酶Q10(CoQ10)和铁抑素处理可以挽救ROS的产生,但TBK1抑制剂GSK-8612则不能(图5J,S7G-J)。因此,STING通过GPX4的自噬降解干扰炎症神经元的适应性抗氧化反应。由于Gpx4缺失导致铁死亡激活,作者假设STING在炎症诱导的神经元铁死亡中发挥关键调节作用。在WT(图5K-L,S7K-L)和Stim1缺失神经元(图S7M-O)中,用抗氧化剂去铁胺、CoQ10和铁抑素处理可保护表达STING的神经元免受谷氨酸兴奋毒性的影响。因此,STING是神经炎症中神经元自噬依赖性铁死亡的关键调节因子。
由于无法确定是否需要稳态cGAS活性来产生低水平cGAMP,从而持续驱动STING运输和信号传导,作者探索了cGAS是否也参与谷氨酸诱导的STING介导的神经元铁死亡。作者构建了缺失Cgas的神经元(图S8A),并通过慢病毒转染或慢性暴露于IFNγ诱导STING表达。在表达STING的神经元中,谷氨酸兴奋毒性诱导的自噬通量增加(图S8B)、GPX4降解(图S8C-D)和ROS产生(图S8E)在WT和缺失Cgas的神经元中是相似的。不表达STING的cGAS缺陷神经元对谷氨酸兴奋毒性的敏感性较低,表明在没有STING的情况下,cGAS在神经元中具有明显的生理作用(图S8F)。因此,由谷氨酸兴奋性毒性引起的STING诱导的铁死亡可能不依赖于神经元中的cGAS。
图5 STING调节炎性神经元的铁死亡
图S6 STING不调节ACSL4、FSP1和SLC7A11的表达
图S7 神经元STIM1缺失增加STING激活的铁死亡
图S8 STING诱导的铁死亡与神经元中的cGAS无关
7.STING抑制改善炎症诱导的神经变性
然而,正如STING能诱导小胶质细胞强激活那样,作者发现经治疗的EAE小鼠中激活的小胶质细数量减少(图S9H)。为了绕过STING拮抗剂对小胶质细胞活化的抑制作用,作者在神经元特异性的Sting-cKO小鼠上构建了EAE。结果表明Sting-cKO小鼠病程改善(图6F,S9I-J),神经元数量增加(图6G),免疫荧光结果表明神经元LC3(图S9K)和4-HNE(图6H)表达下调,GPX4水平上调(图6I)。且没有观察到WT和Sting-cKO EAE小鼠小胶质细胞和CNS浸润免疫细胞的数量存在差异(图S9L,S10A-B)。总的来说,作者的数据表明抑制STING是在神经炎症条件下直接干预NISR的杰出治疗范例,发挥神经保护作用。
图S9 STING增加EAE的神经元自噬
图6 STING抑制剂可防止炎症诱导的神经变性
图S10 WT和神经元Sting-cKO小鼠在EAE过程中表现出相似的免疫细胞浸润和激活
