

Nat Commun:Mtfp1消融可增强线粒体呼吸防止肝脏脂肪
时间 : 2024-01-14代谢功能障碍相关性脂肪肝(MASLD),也被称为NAFLD(非酒精性脂肪肝病),是最常见的慢性肝病,其发病率在全球范围内迅速增长。MASLD是2型糖尿病和肥胖症的常见并发症,其患病率在一般人群中约为30%,在肥胖人群中约为80%。MASLD包括一系列病理学特征,从肝细胞中单纯的脂肪变性,甘油三酯积聚;到代谢功能障碍相关的脂肪性肝炎(MASH);可进一步发展为肝硬化和肝细胞癌(HCC)。
近几十年来,研究人员已经做出巨大努力来研究MASLD进展的机制和治疗靶点。MASLD的进展目前可以用“多重平行撞击”假说来解释,该假说描述了来自不同肝细胞类型的多种事件的协同作用。在肝细胞中,氧化应激和线粒体功能障碍被认为是导致肝细胞损伤和死亡、组织炎症和纤维化的原因之一。线粒体是细胞内重要的细胞器,在细胞内稳态过程中发挥着重要作用。通过氧化磷酸化(OXPHOS)产生三磷酸腺苷(ATP),线粒体也通过隔离和释放促凋亡因子和促炎因子来调节细胞程序性死亡和炎症。然而,线粒体哪一种功能直接涉及脂肪变性、炎症、肝细胞死亡和随后的组织重塑尚未确定。
多项研究报道了线粒体与肝功能的相关性。线粒体基因突变表现为肝功能障碍的遗传性疾病,而线粒体功能障碍与获得性肝病如MASLD,病毒性肝炎和缺血性肝损伤之间也存在强烈的相关性。据报道,线粒体呼吸在人类肝功能障碍进展过程中下降,在MASH患者的肝活检中显示了线粒体结构的扰动,这支持了维持线粒体完整性对肝功能至关重要的观点。肝脏中靶向解偶联剂增加的耗氧量减少了啮齿动物和灵长类动物肝脏脂质积累和脂肪变性的有害作用,然而更广泛范围的线粒体解偶联对其他器官有灾难性的影响。作者最近发现心脏线粒体中的线粒体解偶联受到线粒体分裂过程1(MTFP1)的调节,揭示了这种蛋白质在生物能量调节中的额外作用。MTFP1是一种定位于内膜(IMM)的蛋白质,其名称来源于体外线粒体分裂的公认作用,并已引起人们的兴趣,成为人类肝功能障碍的标志物。有研究揭示了肿瘤组织中MTFP1的表达与HCC患者生存之间的联系,其发展受到MASLD进展的严重影响。暗示MTFP1在体外代谢感应和细胞程序性死亡调节中的作用,但其在肝功能和代谢中的相关性从未被探索过。
2023年12月20日,法国巴黎西岱大学Timothy Wai研究团队在Nature Communications发表了题为“Mtfp1 ablation enhances mitochondrial respiration and protects against hepatic steatosis”的研究论文。研究报道,当小鼠喂食高脂饮食(HFD)时,体内肝细胞中Mtfp1的缺失增强了肝OXPHOS活性并减弱了饮食诱导的肝脂肪变性,体重增加和全身葡萄糖失调。
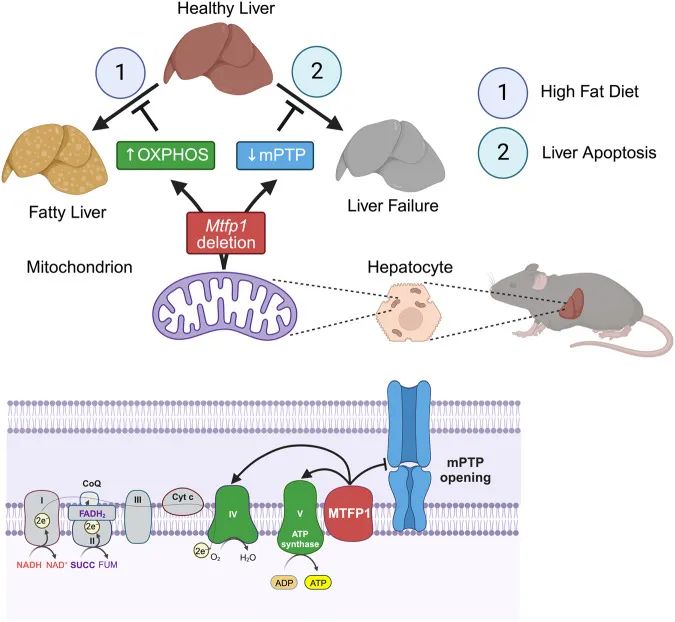
图解摘要

“
肝脏特异性Mtfp1敲除小鼠模型的建立
为了研究线粒体功能与肝脏代谢之间的联系,作者构建了肝脏特异性KO小鼠模型,特异性敲除了肝细胞(图1A)中的Mtfp1。将先前在C57Bl6/N背景上产生的条件小鼠(Mtfp1LoxP/LoxP)与Alb-Cre重组酶转基因小鼠(Alb-Cretg/+)杂交以产生LMKO小鼠(Alb-Cretg/+ Mtfp1LoxP/LoxP)。LMKO小鼠中Mtfp1的敲除是肝脏特异性的(图1B),肝细胞中的MTFP1消融不影响围产期存活(图1E),对LMKO小鼠肝脏进行的组织学分析与对照同窝小鼠无显著差异(图1F)。与心脏中MTFP1的消融相反,在正常饮食(NCD)下,LMKO小鼠没有表现出任何明显的缺陷。对肝脏结构和功能的综合评估显示没有缺陷:LMKO小鼠的肝脏质量(图1G)没有变化,肝损伤的循环生物标志物如丙氨酸氨基转氨酶(ALAT)和天冬氨酸氨基转氨酶(ASAT)的水平相对于对照同窝动物没有增加(图1H)。循环中胆固醇和甘油三酯水平正常(图1I),代谢性能没有受损(图1J)。对照和LMKO肝脏几乎没有基因表达失调。综上所述,结果表明,在基础条件下,肝细胞中Mtfp1的缺失不会对肝脏产生负面影响。
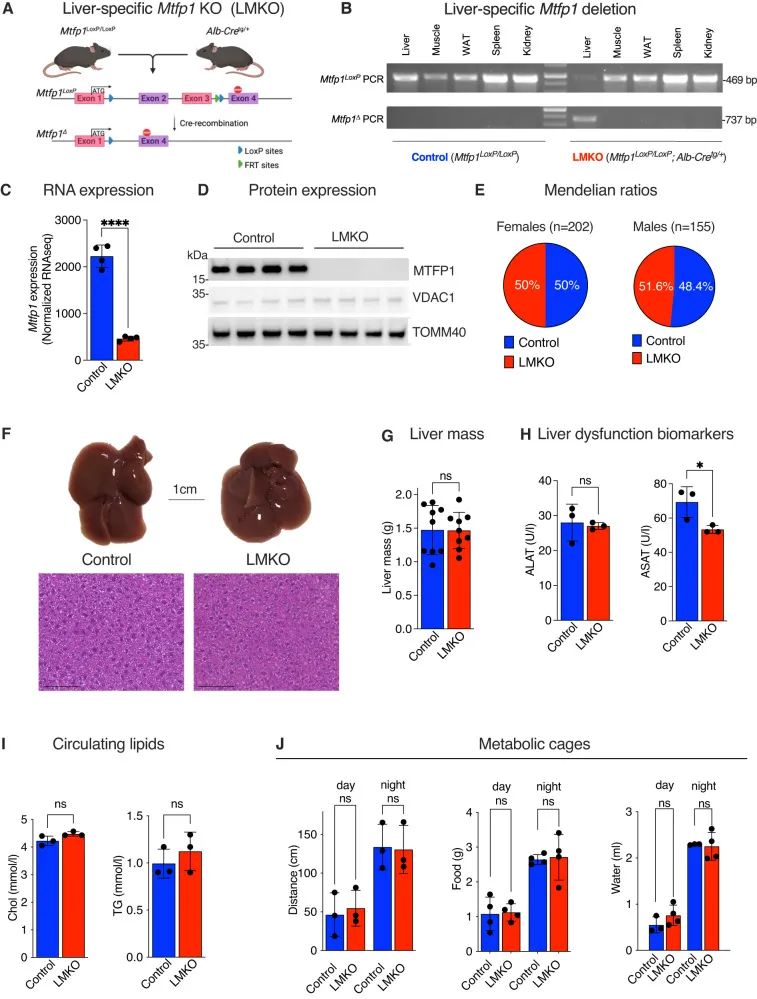
图1 Mtfp1在小鼠肝脏中的缺失不会损害基础肝功能
“
Mtfp1缺失增加OXPHOS活性和线粒体呼吸
根据之前的研究发现MTFP1消融降低了心脏线粒体的生物能量效率,作者直接评估Mtfp1缺失对肝脏线粒体生物能量学的影响。为此,对从对照和LMKO小鼠分离的肝线粒体进行了高分辨率的荧光呼吸测定,通过Rhodamine 123染色(RH-123)测量了耗氧率(JO2)和线粒体膜电位(ΔΨ)变化。JO2和RH-123记录了线粒体与呼吸底物温育促进电子传递至复合物I(状态2;丙酮酸,谷氨酸和苹果酸(PGM)或复合物II(状态2;琥珀酸和鱼藤酮),或在棕榈酰肉碱加苹果酸(状态2)在磷酸化(状态3:ADP)和非磷酸化(状态4:寡霉素(OLGM)抑制ATP合酶)状态(图2A)。与MTFP1缺陷的心脏线粒体相反,在测试的任何呼吸底物存在下,LMKO肝线粒体中的呼吸在磷酸化(状态3)条件下显著增加。丙酮酸、谷氨酸和苹果酸导致状态3呼吸增加49%,琥珀酸和鱼藤酮导致呼吸增加57%(图2B)。在脂肪酸酯衍生物棕榈酰肉碱存在下,状态3呼吸增加200%,提示由肝脏特异性缺失Mtfp1引起的脂肪酸衍生的能量代谢效率增加。
此外,在棕榈酰肉碱加苹果酸存在下,LMKO肝线粒体显示出较高的呼吸控制比(RCR)(图2C)。尽管状态3呼吸显著增加,但作者没有观察到线粒体膜电位的基因型特异性差异(图2D),因为增加的耗氧率通常伴随着由于通过复合物V(合成ATP)消散的质子动力而导致的膜电位减少。对这一结果最简单的解释是,MTFP1消融促进了细胞色素c氧化酶(复合体IV)和ATP合酶(复合体V)活性的相应增加。当作者在单独的测定中测量复合物IV(图2E)和复合物V(图2F)的特异性活性时,发现相对于同窝对照,LMKO线粒体增加约20%,表明两种复合物在增加呼吸同时维持线粒体膜电位。作者进一步证实了这个发现,通过测量用NADH,Cyt c,琥珀酸盐和鱼藤酮提供的丝裂原体中的耗氧速率来驱动通过复合物II或NADH,Cyt c,琥珀酸盐和丙二酸盐通过复合物I驱动电子转运(图2G)。在两种测定中,LMKO肝线粒体的耗氧量都升高,表明复合物IV的活性在MTFP1消融时独立于原动力而本质上增加。总之,这些观察结果提示MTFP1消融触发OXPHOS机制组分的特异性上调或诱导一般线粒体生物合成反应。为了区分这些可能性,作者使用多分子和基于成像的方法评估线粒体质量。定量从对照或LMKO小鼠分离的原代肝细胞线粒体表达基因编码靶向YFP(mitoYFP)的基质显示荧光信号强度或表面积没有差异。表明在原代肝细胞中线粒体不受MTFP1消融的影响。肝组织中线粒体DNA含量的定量显示在喂养NCD的小鼠中没有特异性差异。肝切片的S2D和透射电镜(TEM)分析显示,对照组和LMKO小鼠之间的线粒体面积和线粒体长度没有差异,进一步排除对线粒体质量和形态的影响。调节线粒体动力学的关键蛋白水平在LMKO小鼠中保持不变和DRP1的线粒体定位一样,进一步排除了对线粒体动力学的影响。总之,数据表明,肝细胞中复合体IV和V活性的特异性和协调性增加增强了Mtfp1缺失的肝线粒体的呼吸作用,使其能够以较高的速率消耗呼吸底物。
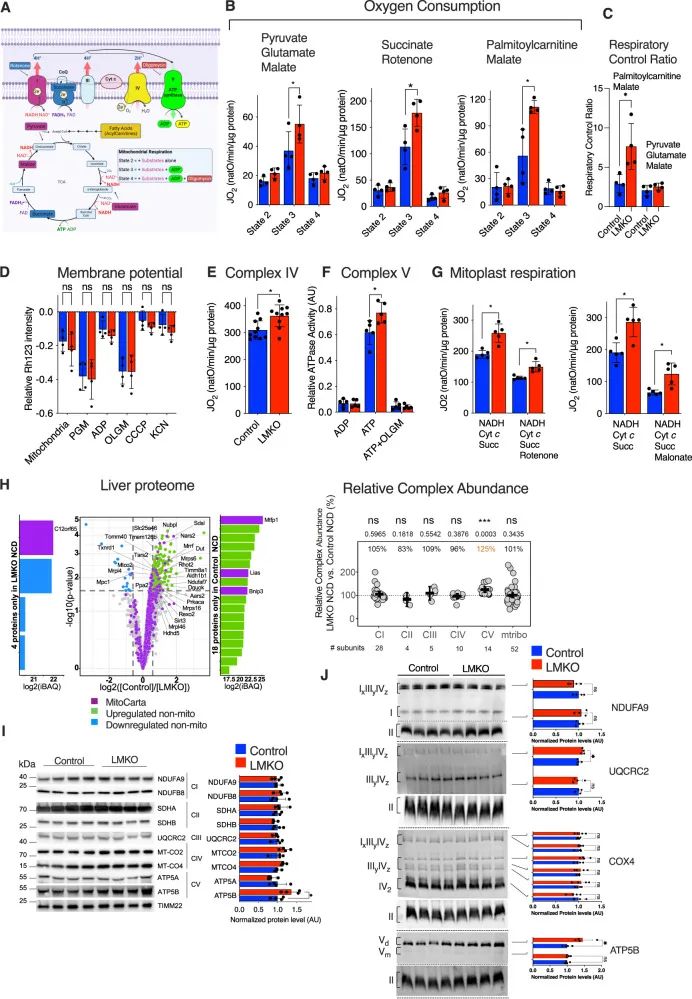
图2 Mtfp1缺失通过OXPHOS上调促进线粒体呼吸
“
MTFP1与肝脏中OXPHOS相关蛋白相互作用
为了深入研究增加特异性活性的机制,作者在NCD喂养的LMKO小鼠的肝脏中通过蛋白质组学定量的线粒体蛋白质来评估相对复合物丰度(RCA)。这些数据显示复合体Ⅴ亚基显著增加,通过定量SDS-PAGE(图2I)和蓝色天然聚丙烯酰胺凝胶电泳(BN-PAGE)分析(图2J)证实。LMKO肝线粒体中OXPHOS复合物的稳态水平的BN-PAGE分析显示复合物V二聚体的增加(图2J)与报道的ATP酶活性增加一致(图2F)。综上所述,数据表明,复合物V的组装和维持的改善以及复合物IV活性的增加是LMKO肝线粒体中观察到的呼吸增强的原因。
为了进一步研究MTFP1与线粒体内膜(IMM)中大分子复合物之间的关系,作者试图评估MTFP1在肝脏中的相互作用。作者构建了肝特异性转基因小鼠模型,使得能够从Rosa26基因座(以下称为肝细胞FLAG-MTFP1小鼠; 图3A)表达FLAG-MTFP1。作者通过蛋白酶保护试验证实肝细胞FLAG-MTFP1小鼠在IMM中正确表达FLAG-MTFP1(图3B)。然后,将肝线粒体进行免疫共沉淀(Co-IP)和质谱法(MS/MS)以定义相互作用的蛋白质伴侣,这使得能够通过MS/MS分析(图3C,D,补充数据集3)鉴定MTFP1的112个特异性相互作用物(倍数变化> 2)。然后,作者决定将该列表细分为二元相互作用者和富集相互作用者:二元相互作用者是指只能在肝细胞FLAG-MTFP1而非对照组肝线粒体CoIP洗脱液中识别的蛋白质,而富集相互作用者是指在肝细胞FLAG-MTFP1小鼠的CoIP洗脱液中肽丰度较高的蛋白质(图3D)。
二元相互作用蛋白的分类揭示了广泛的线粒体功能,与线粒体翻译和OXPHOS有关的因子显著丰富(图3D)。另一方面,富集的相互作用物没有显示线粒体翻译所需的蛋白质的富集,而是那些参与线粒体内各种代谢过程的蛋白质,包括SLC25A家族的载体蛋白。发现SLC25A22,SLC25A11,SLC25A20和SLC25A15与肝脏中的MTFP1相互作用(图3D),但非靶向代谢组学分析显示这些载体特异性调节的代谢物没有改变。二维-BN-PAGE分析显示,肝线粒体中形成了MTFP1大分子复合物,而LMKO小鼠体内不存在这种复合物,并且这种复合物与SLC25A4(ANT1)共同迁移。(图3E)总之,这些数据表明线粒体基因表达和IMM复合物丰度和活性的转录后调节通过肝细胞中MTFP1的缺失以特定的方式诱导,导致线粒体呼吸增强。
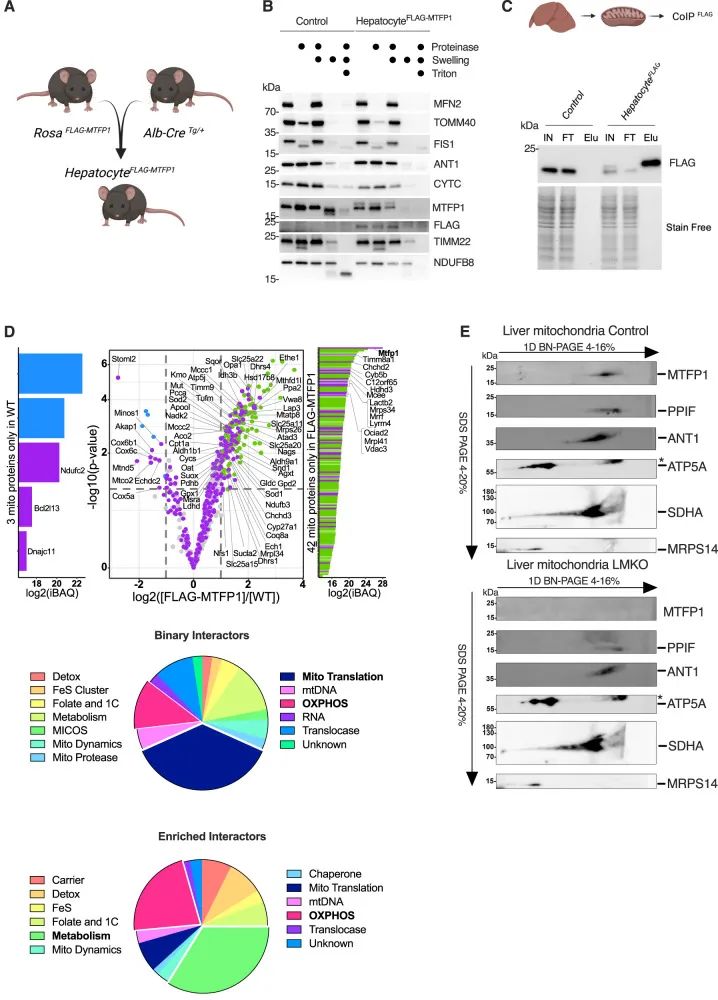
图3 MTFP1与肝脏中各种IMM成分相互作用
“
Mtfp1基因缺失对高脂饮食诱导的肝脂肪变性的保护作用
鉴于LMKO肝线粒体的刺激性呼吸活性增强,作者接下来决定评估LMKO小鼠对高脂饮食(HFD)喂养的反应。给对照组和LMKO小鼠喂食HFD,从8周龄开始,持续16周,之前已经证明这可以驱动饮食诱导的肥胖和MASLD。在对照小鼠中,观察到HFD喂养引发肝脏肥大(图4A),HFD喂养还引起体重增加(图4C)和肝脏甘油三酯(TG)的积累(图4D),组织学分析显示脂滴大量积聚(图4E)。相比之下,HFD喂养的LMKO小鼠免于体重增加(图4C)和肝功能障碍,与对照相比,显示肝脏质量减少26%(图4A,B)。一致的是,来自HFD喂养的LMKO小鼠的肝脏的组织学分析显示脂肪变性减少(图4E),肝TG积累完全恢复到在NCD处理的小鼠中测量的水平(图4D),并且ALAT降低64%和ASAT降低32%(图4F)。在LMKO小鼠中对HFD喂养的代谢保护反映在分子水平上:肝RNAseq分析显示HFD饮食会诱导对照组肝脏出现明显的转录失调,其中854个基因上调,758个基因下调(图4G)。GO分析表明,对照组小鼠因HFD饮食而导致各种代谢途径失调,包括脂质和支链氨基酸代谢、PPAR信号转导、类固醇生物合成和脂肪酸代谢(图4H)。这些改变在HFD喂养的LMKO小鼠中不存在,其中仅检测到214个上调基因和164个下调基因,其中大部分涉及应激反应和免疫信号传导途径(图4H)。在蛋白质水平,通过肝脏蛋白质组学分析,观察到在对照小鼠中HFD饮食诱导复合物IV的全面减少。这种反应在LMKO小鼠中减弱,HFD饮食的LMKO小鼠复合物IV蛋白的相对增加。
鉴于缺失Mtfp1时在体内和体外观察到的对肝细胞和肝脏损伤的保护,作者探究对HFD诱导的肝脏代谢失调的保护是否可以通过对细胞死亡的不同敏感性来解释。然而,在来自HFD喂养的对照和LMKO小鼠的组织切片上进行的TUNEL测定显示不存在肝细胞凋亡(图4I),这与之前HFD引起有限的肝细胞死亡的发现一致。总之,数据表明,肝细胞中的Mtfp1缺失防止了体内肝脂肪变性,其方式与抗凋亡无关。
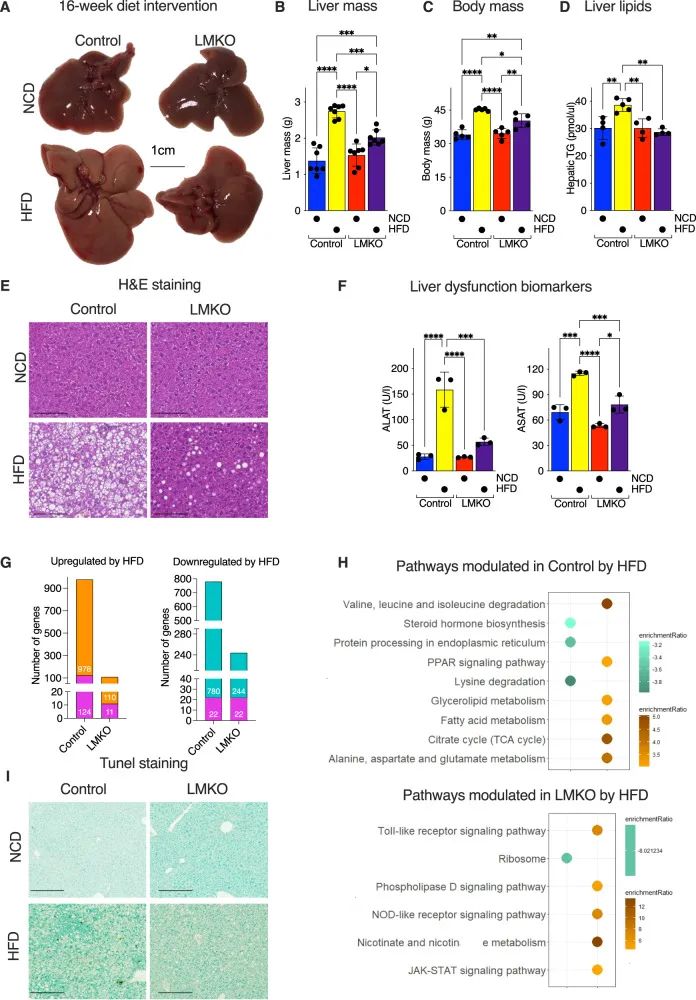
图4 肝脏Mtfp1基因缺失对高脂饮食诱导小鼠脂肪变性的保护作用
“
Mtfp1缺失以细胞自主方式改善肝细胞代谢
在证明LMKO小鼠能防止饮食诱导的脂肪变性之后,作者接下来试图通过在HFD喂养后对对照和LMKO小鼠进行一系列代谢测试来确定保护作用。脂肪堆积已知会影响全身葡萄糖稳态。与肝细胞中MTFP1消融促进的肝代谢改善一致,代谢笼分析显示喂食HFD的LMKO小鼠的全身呼吸得到改善。在LMKO小鼠中观察到的较低的呼吸交换率(RER)(图5A),LMKO小鼠比对照同窝动物更有效地代谢营养来源的脂质,这很可能归因于这些小鼠肝脏中的内在差异。对照组和LMKO小鼠的食物和水摄入量没有改变。已知由HFD喂养引起的代谢失调增加了啮齿动物肝脏的葡萄糖从头合成,因此作者试图通过腹膜内丙酮酸耐受试验(IP-PTT)来评估糖异生。观察到HFD喂养的对照小鼠相对于NCD同窝仔鼠的血糖水平增加了1.5倍,在HFD喂养的LMKO组中降低了14%(图5B),表明肝细胞中的Mtfp1缺失保护肝脏免受饮食诱导的糖异生失调。与这些发现一致,腹膜内葡萄糖耐量试验(IP-GTT)显示LMKO小鼠适度地保护HFD诱导的葡萄糖耐受不良。在对照小鼠中,HFD喂养导致IP-GTT的曲线下面积(AUC)相对于正常食物饮食(NCD)对照增加1.7倍, HFD喂养的LMKO小鼠减少17%(图5C)。为了排除HFD喂养的LMKO小鼠的葡萄糖耐量改善是胰岛素抵抗改变的结果,而不是糖异生改善,作者进行了腹腔内胰岛素耐量试验(IP-ITT)。观察到HFD或NCD的胰岛素敏感性没有差异(图5D),表明LMKO小鼠的葡萄糖耐量改善不是由全身胰岛素敏感性增加引起的。一致的是,作者没有观察到对照和喂食NCD的LMKO小鼠之间基础胰岛素水平的差异,并且用胰岛素刺激的禁食LMKO小鼠的肝胰岛素信号传导没有差异。虽然LMKO小鼠在HFD喂养后体重增加较少(图4C),但是通过NMR微型分析(图5E)和胆固醇和甘油三酯的循环水平(图5F)没有观察到体重差异。根据这些发现,LMKO小鼠没有减少肝外脂肪堆积(图5G)、白色脂肪组织堆积和脂肪细胞肥大(图5)。作者也没有观察到来自LMKO小鼠的褐色脂肪组织中Ucp1和Prdm16表达的差异,表明BAT功能不太可能被肝脏特异性缺失的Mtfp1间接影响。总之,数据表明,通过肝脏特异性缺失Mtfp1而赋予小鼠的对饮食诱导的代谢失调的保护作用主要局限于肝脏。
为了确定肝细胞缺失Mtfp1是否以细胞自主的方式防止脂肪变性,作者建立了一种测定方法,从NCD喂养的小鼠分离的原代肝细胞模拟HFD诱导的脂肪变性。将原代肝细胞铺板并在脂质复合物(IntLip)存在下培养:由亚油酸,油酸,棕榈酸和硬脂酸组成的复合脂质乳剂,这是HFD中发现的最丰富的脂肪酸。从NCD饮食的对照小鼠分离的原代肝细胞的IntLip处理24小时后导致细胞内脂质积累增加1.6倍,通过BODIPY荧光活细胞成像(图5H)和高通量共聚焦成像进行定量(图5I)原代肝细胞中Mtfp1的缺失保护了了这种表型。因此,得出结论,肝细胞中Mtfp1的缺失通过以细胞自主的方式改善肝细胞代谢,直接保护肝脏免受HFD诱导的代谢失调。
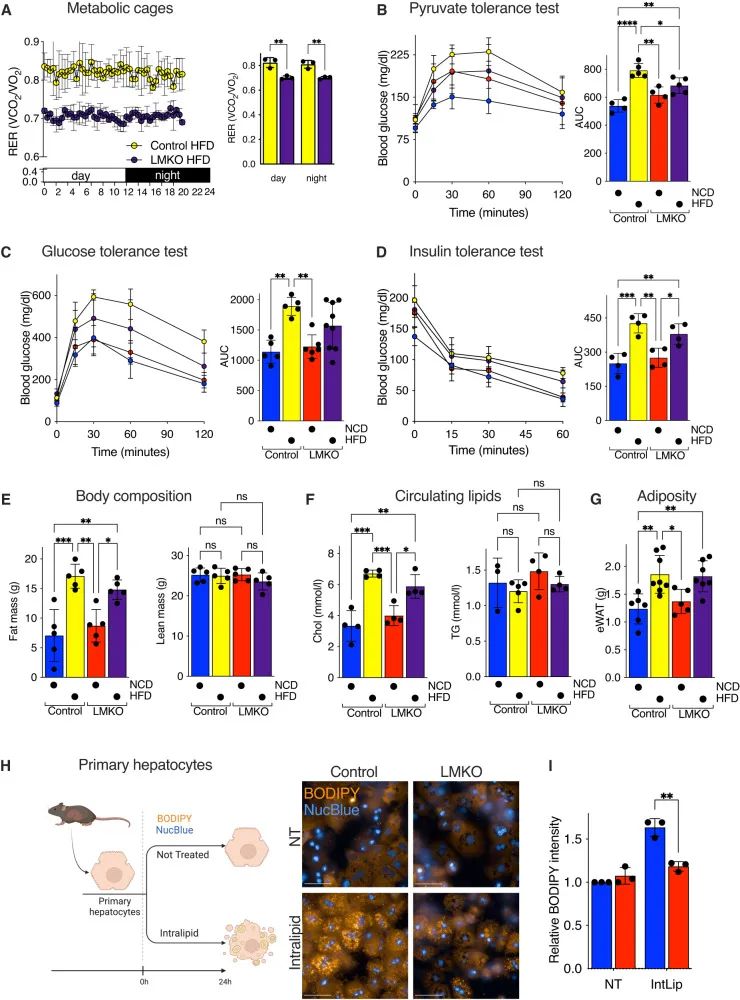
图5 LMKO小鼠受到饮食诱导的代谢失调的保护
“
Mtfp1缺失对肝细胞死亡的保护作用
鉴于MTFP1在各种细胞系中报道的细胞死亡敏感性的影响以及MTFP1与人类肝肿瘤进展的关联,作者试图直接测试MTFP1是否参与体内细胞存活的调节。通过用FAS抗体处理3个月大的小鼠24小时来测量对肝细胞凋亡的反应,FAS抗体通过激活FADD信号通路诱导凋亡性肝脏损伤,其方式受IMM完整性的调控。用FAS处理的WT小鼠的肝脏显示出纤维化和坏死病灶(图6A)。在对照肝脏中,ALAT,ASAT和乳酸脱氢酶(LDH)(一种更普遍的组织损伤标志物)分别显著增加了35,85和45倍(图6B)。与FAS处理的对照小鼠相比,LMKO肝脏的H&E结果显示减弱了FAS诱导的损伤(图6C),ALAT减少84%,ASAT减少86%,LDH减少97%(图6B)。TUNEL染色分析显示,与对照小鼠相比,FAS处理的LMKO小鼠的细胞死亡显著减少(图6D)。综上所述,这些研究表明肝脏特异性Mtfp1消融可以在体内保护细胞凋亡诱导的肝脏损伤。
为了研究MTFP1消融是否以细胞自主的方式保护肝细胞免于细胞死亡,作者在对照和LMKO小鼠中分离原代肝细胞,并在体外进行细胞死亡测定。为了诱导细胞死亡,在铺板后16小时用过氧化氢(H2O2 1mM)处理原代肝细胞,通过碘化丙啶(PI)摄取动力学监测细胞死亡。与在体内的观察一致,与对照原代肝细胞相比,用H2O2离体处理的MTFP1-/-原代肝细胞显著抑制细胞死亡(4小时减少52%)(图6E,F),证明MTFP1消融保护肝细胞死亡。肝脏中的细胞死亡,特别是肝细胞中的细胞死亡,可以通过线粒体通透性转换孔(mPTP)的开放来严格调节。为了测试Mtfp1缺失是否影响mPTP开放,作者分离了肝线粒体并模拟了氯化钙超载的mPTP开放(图6G)。与对照同窝鼠相比,观察到从LMKO小鼠分离的肝线粒体中mPTP开放的敏感性降低(图6G),其可以通过用mPTP抑制剂环孢素A(CsA)预处理来消除。综上所述,得出结论,Mtfp1在肝细胞中的缺失对mPTP开放和诱导细胞死亡具有保护作用。
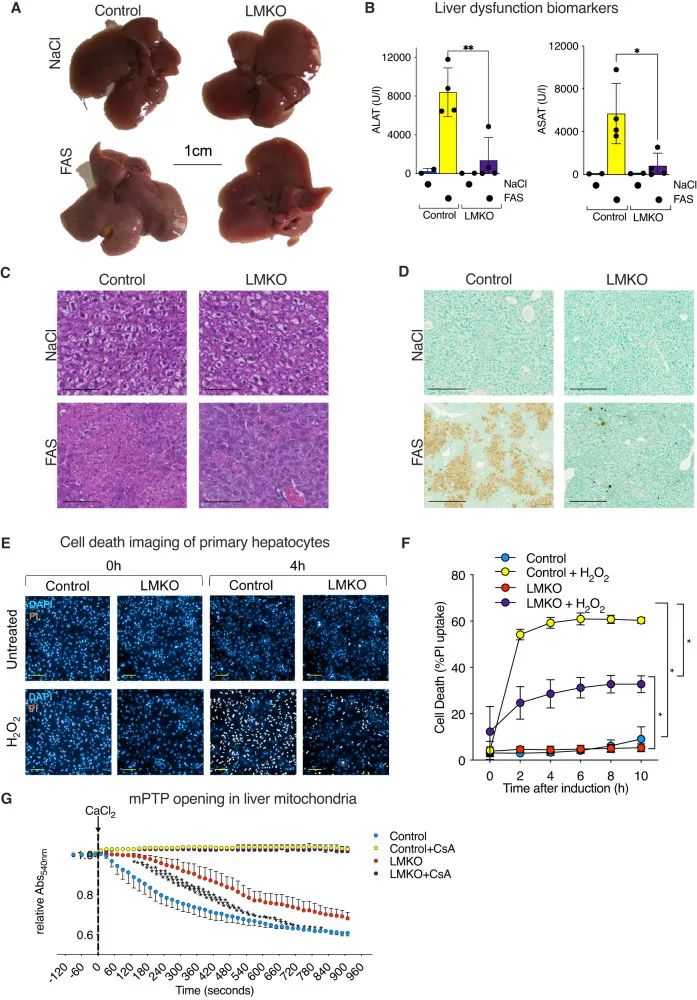
图6 Mtfp1基因缺失对FAS小鼠肝损伤的保护作用
